Diagnosing Fabry Disease in Clinical Practice
.webp1/jcr:content/science%20hero%20(6).webp)
Clinical Manifestations of FD
Classical FD (<3% enzyme activity)
 |
2nd decade |
 |
Symptoms appear later. |
 |
Neuropathic pain, angiokeratomas, and/or cornea verticillata are absent in females. |
 |
Childhood and adolescence |
 |
Neuropathic pain |
 |
Hypohidrosis and hyperhidrosis |
 |
Febrile crisis |
 |
Eye involvement* |
 |
Hearing loss |
 |
Angiokeratoma |
 |
Microalbuminuria |
 |
GI symptoms |
 |
2nd decade |
 |
Cardiomyopathy |
 |
Stroke and TIA |
 |
Macroproteinuria and eGFR loss |
 |
From 3rd decade |
 |
Progressive organ damage |
 |
Organ failure |
 |
Premature death |
 |
Nonclassical or late-onset FD (3%–30% enzyme activity) |
| Variable disease course and single organ involvement | |
| Cardiac variant is common in late-onset FD | |
| Identified in patients with stroke, renal failure, or cardiomyopathy | |
| No neuropathic pain, angiokeratomas, and/or cornea verticillata |
 |
FD diagnosis requires a multidisciplinary team approach involving: |
|
Biochemist |
Pediatrician |
Neurologist |
Cardiologist |
Dermatologist |
Nephrologist |
Geneticist |
 |
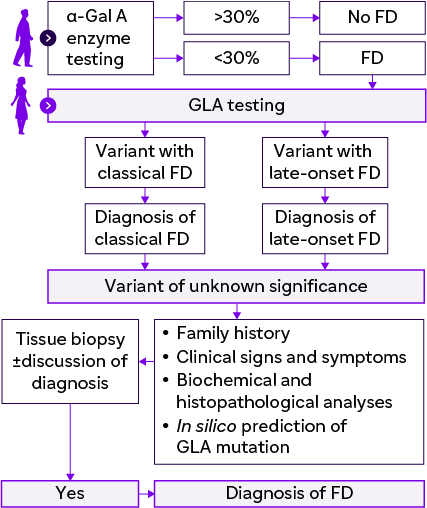
Suggested diagnostic algorithm for patient with clinically suspected FD |
 |
Testing for FD |
1. Lyso-Gb3 indicates severity of FD
2. Endomyocardial and renal biopsies are used
3. Genetic testing
 |
Testing for FD
|
 |
LSD referral centers to manage FD from diagnosis to long-term follow-up |
 |
FD: |
Rare but underdiagnosed |
To be known and recognized by internal medicine physicians |
Early treatment can change the natural course of the disease. |
*Eye involvement includes cornea verticillata, tortuous retinal vessels, cataracts, and conjunctival lymphangiectasia.
Rare but To be known and recognized by underdiagnosed internal medicine physicians α-Gal A: Alpha-galactosidase; eGFR: Estimated glomerular filtration rate; GLA: α-Galactosidase A; FD: Fabry disease; GI: Gastrointestinal; LSD: Lysosomal storage disease; Lyso-Gb3: Globotriaosylsphingosine; TIA: Transient ischemic attack.


