
Mozobil® (plerixafor)
Mozobil contiene el principio activo plerixafor, antagonista selectivo reversible del receptor de quimiocina CXCR4 que bloquea la unión de su ligando afín, el factor derivado de células estromales 1α (SDF-1α), también conocido como CXCL12. Esta acción permite la liberación de células madre de la médula ósea a la sangre periférica.1

Indicaciones
Mozobil está indicado, en combinación con un factor estimulante de colonias de granulocitos (G-CSF), para potenciar la movilización de células madre hematopoyéticas a sangre periférica para su recogida y posterior trasplante autólogo en pacientes adultos con linfoma o mieloma múltiple cuyas células se movilicen con dificultad.1
Pacientes pediátricos (de 1 a menos de 18 años) en combinación con G-CSF para potenciar la movilización de células madre hematopoyéticas a sangre periférica para su recogida y posterior trasplante autólogo en niños con linfoma o tumores sólidos malignos, ya sea:
- de forma preventiva, cuando se considera que el recuento de células madre circulantes en el día previsto de recogida, después de la movilización adecuada con G-CSF (con o sin quimioterapia), es insuficiente respecto al rendimiento deseado de células madre hematopoyéticas, o
- cuando no se logra recoger de forma previa suficientes células madre hematopoyéticas.1
Administración y dosis de Mozobil®
Debe administrarse mediante inyección subcutánea en un plazo de 6 a 11 horas antes de iniciar cada aféresis y después de pretratamiento con G-CSF de 4 días de duración. En los ensayos clínicos, con frecuencia se ha utilizado Mozobil de 2 a 4 (y hasta 7) días consecutivos.1
La dosis diaria recomendada de plerixafor por inyección subcutánea (SC) es:
-
dosis fija de 20 mg o 0,24 mg/kg de peso corporal para pacientes con un peso ≤ 83 kg
-
0,24 mg/kg de peso corporal para pacientes con un peso > 83 kg.
La dosis diaria recomendada de plerixafor por inyección subcutánea (SC) es: 0,24 mg/kg de peso corporal.
En función del aumento de exposición con el aumento del peso corporal, la dosis de plerixafor no debe superar los 40 mg/día.1
En los pacientes con aclaramiento de creatinina de 20 a 50 ml/min se debe reducir la dosis de plerixafor en un tercio hasta 0,16 mg/kg/día. No existe experiencia clínica suficiente para hacer recomendaciones posológicas alternativas ni para pacientes con un aclaramiento de creatinina < 20 ml/min, ni para pacientes en hemodiálisis. En función del aumento de exposición con el aumento del peso corporal, la dosis de plerixafor no debe superar los 27 mg/día si el aclaramiento de creatinina es inferior a 50 ml/min.1
El tratamiento con Mozobil debe ser iniciado y supervisado por un médico especialista en oncología y/o hematología.1
Movilización en TAPH
El objetivo principal de la movilización es recoger suficientes células madre CD34+ para que el paciente pueda someterse a TAPH sin demora 2,3

Según el consenso español sobre movilización y recolección de células madre hematopoyéticas en trasplante autólogo:11
-
Pacientes muy pretratados. (Más de tres líneas de tratamiento; incluyendo TAPH previo).
-
Enfermedad avanzada o activa.
-
Edad avanzada. (Más de 60 años).
-
Baja reserva de médula ósea, definida como:
-
Retraso en la recuperación hematopoyética en ciclos de quimioterapia anteriores.
-
Niveles de leucocitos bajos (inferior a 3 × 109/ml), hemoglobina (inferior a 9 g/dl), y especialmente recuento de plaquetas (inferior a 120 × 109/ml) en el hemograma del paciente, antes de la movilización.
-
Tratamiento previo de radioterapia, especialmente aquellos con amplios campos irradiados.
-
Medicamentos de quimioterapia anteriores; especialmente: Fludarabina, Bendamustina o Agentes alquilantes.
-
Tratamiento previo con algunos medicamentos nuevos; especialmente IMiDs: (Lenalidomida o Pomalidomida).
-
Cuando se excluyen complicaciones durante el procedimiento, bajo número de CD34+ recogidas durante la leucaféresis (menos de 2 × 106/kg).
-
La tasa de fracaso de movilización puede alcanzar el 40% con las estrategias de movilización tradicionales 2,*
Consecuencias potenciales al no alcanzar los niveles objetivos de CD34+

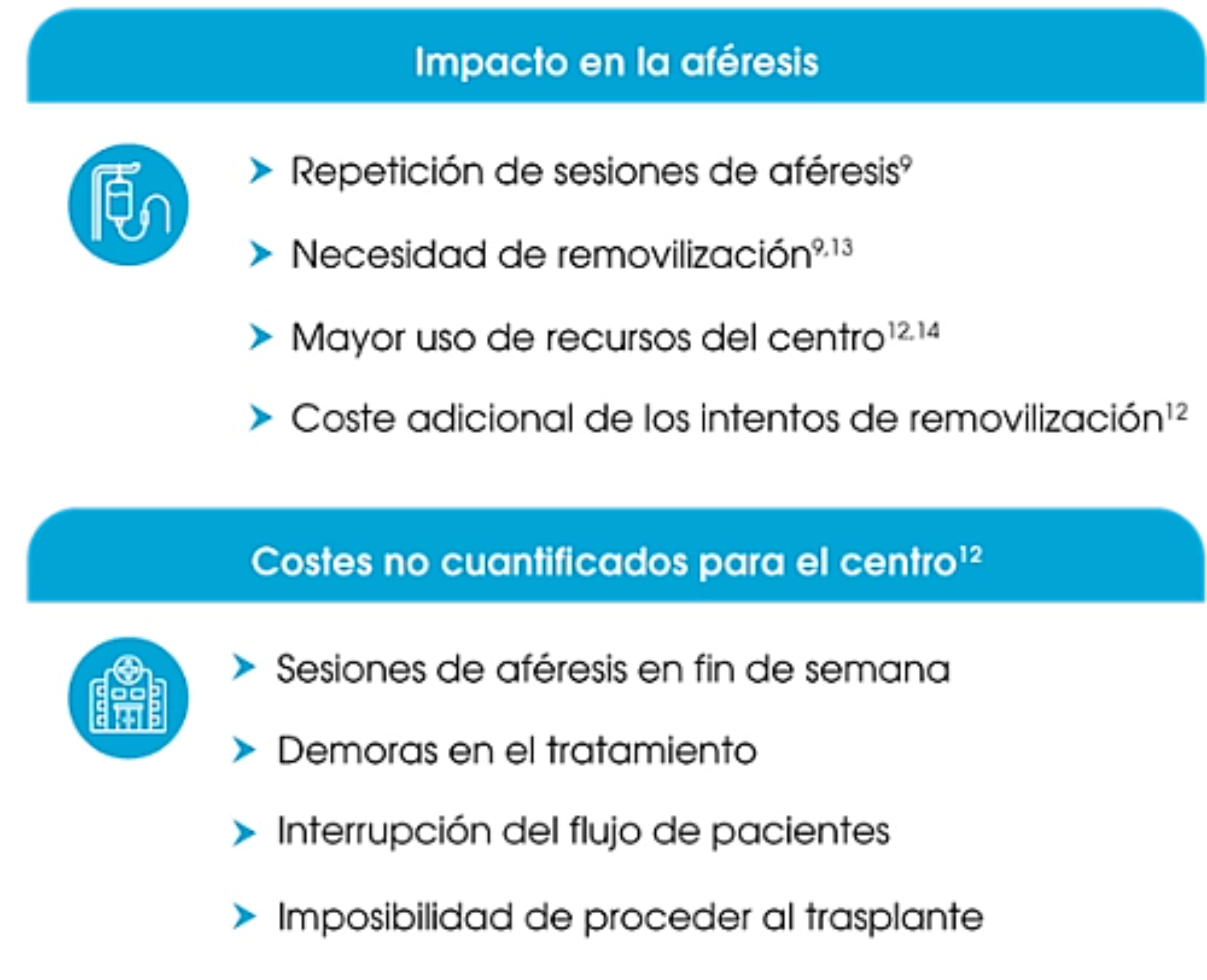
*El fracaso de la movilización fue del 18% al 41% y se definió como un < 2 x 106células CD34+/kg.2
El objetivo principal de la movilización es recoger suficientes células madre CD34+ para que el paciente pueda someterse a TAPH sin demora 2,3
Según el consenso español sobre movilización y recolección de células madre hematopoyéticas en trasplante autólogo:11
-
Pacientes muy pretratados. (Más de tres líneas de tratamiento; incluyendo TAPH previo).
-
Enfermedad avanzada o activa.
-
Edad avanzada. (Más de 60 años).
-
Baja reserva de médula ósea, definida como:
-
Retraso en la recuperación hematopoyética en ciclos de quimioterapia anteriores.
-
Niveles de leucocitos bajos (inferior a 3 × 109/ml), hemoglobina (inferior a 9 g/dl), y especialmente recuento de plaquetas (inferior a 120 × 109/ml) en el hemograma del paciente, antes de la movilización.
-
Tratamiento previo de radioterapia, especialmente aquellos con amplios campos irradiados.
-
Medicamentos de quimioterapia anteriores; especialmente: Fludarabina, Bendamustina o Agentes alquilantes.
-
Tratamiento previo con algunos medicamentos nuevos; especialmente IMiDs: (Lenalidomida o Pomalidomida).
-
Cuando se excluyen complicaciones durante el procedimiento, bajo número de CD34+ recogidas durante la leucaféresis (menos de 2 × 106/kg).
-
La tasa de fracaso de movilización puede alcanzar el 40% con las estrategias de movilización tradicionales 2,*
Consecuencias potenciales al no alcanzar los niveles objetivos de CD34+
*El fracaso de la movilización fue del 18% al 41% y se definió como un < 2 x 106células CD34+/kg.2
Eficacia y seguridad de Mozobil®
Mozobil + G-CSF redujo significativamente el fracaso de movilización en comparación con G-CSF solo 7,8

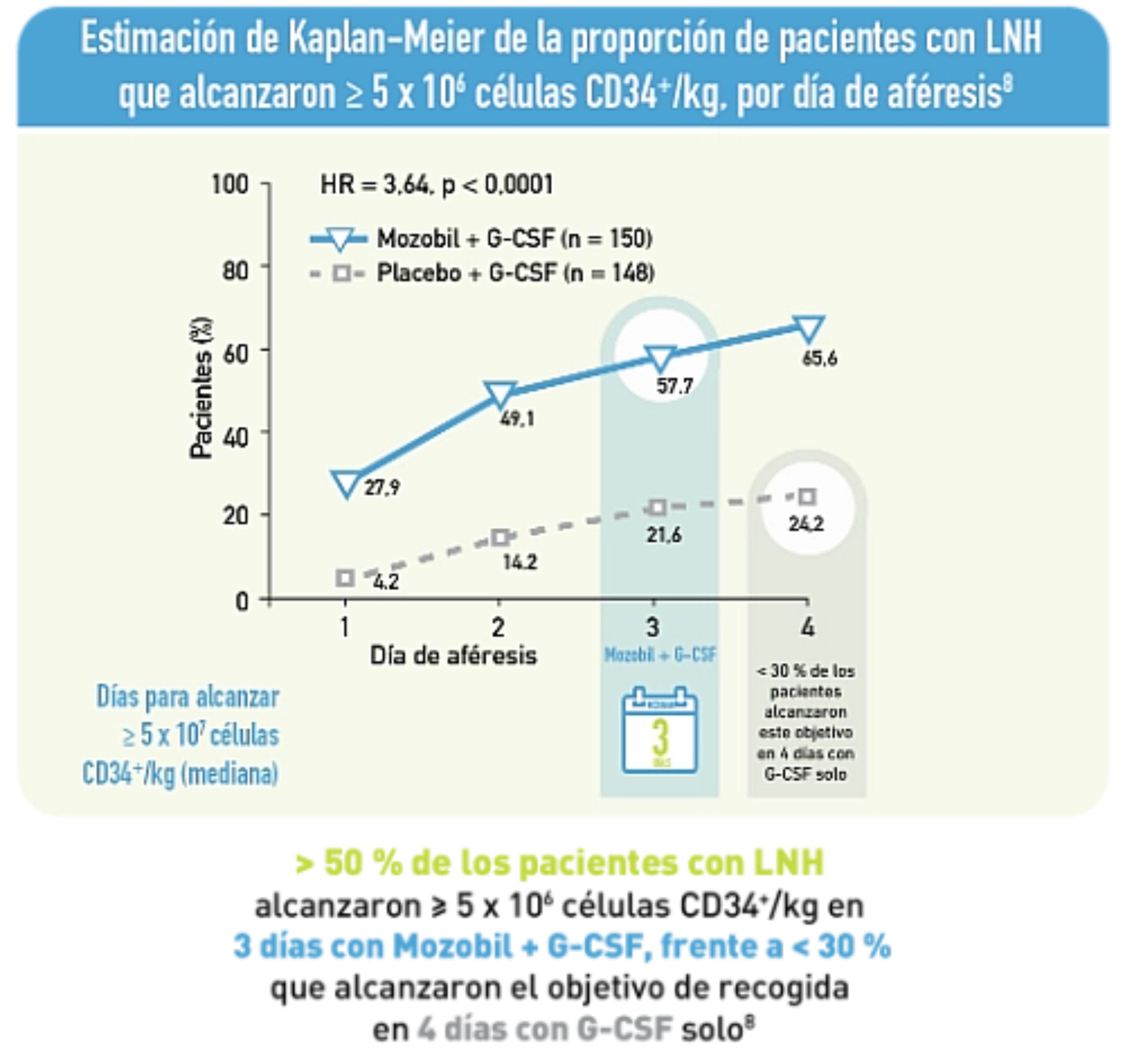
Mozobil + G-CSF redujo el número de días de aféresis 7,8

Mozobil + G-CSF permitió que más pacientes procedieran al TAPH 7,8


Análisis post-hoc de pacientes con MM o LNH que recibieron Mozobil + G-CSF, frente a pacientes que recibieron solo G-CFS, que se sometieron a trasplante después de alcanzar un número mínimo (2x 106células CD34+/kg) de células recogidas.7, 8
Mozobil + G-CSF tiene un perfil de seguridad estable 7,8
Efectos adversos más comunes relacionados con el tratamiento del estudio que ocurrieron en > 5% de los pacientes en cualquier grupo de tratamiento durante el periodo 1

Adaptación de DiPersio JF. et al (2009)7 y DiPersio JF. et al (2009).8
Mozobil + G-CSF redujo significativamente el fracaso de movilización en comparación con G-CSF solo 7,8
Mozobil + G-CSF redujo el número de días de aféresis 7,8
Mozobil + G-CSF permitió que más pacientes procedieran al TAPH 7,8
Análisis post-hoc de pacientes con MM o LNH que recibieron Mozobil + G-CSF, frente a pacientes que recibieron solo G-CFS, que se sometieron a trasplante después de alcanzar un número mínimo (2x 106células CD34+/kg) de células recogidas.7, 8
Mozobil + G-CSF tiene un perfil de seguridad estable 7,8
Efectos adversos más comunes relacionados con el tratamiento del estudio que ocurrieron en > 5% de los pacientes en cualquier grupo de tratamiento durante el periodo 1
Adaptación de DiPersio JF. et al (2009)7 y DiPersio JF. et al (2009).8
En pacientes con MM o LNH, Mozobil + G-CSF permite:7,8
Reducir el número de días de aféresis
Reducir el fracaso de movilización
Mayor número de pacientes proceden al TAPH
Reducir el número de días de aféresis
Reducir el fracaso de movilización
Mayor número de pacientes proceden al TAPH
Movilización de Células Progenitoras Hematopoyéticas
MONOGRAFÍA: CASOS CLÍNICOS
Coordinación: Dr. Rafael Duarte Palomino | Dr. José Luis Bueno Cabrera
Se presentan en esta monografía tres casos clínicos de uso de Mozobil.

Publicaciones

Estudio fase I/II de dos brazos (MOZAIC): Plerixafor en pacientes pediátricos con tumores sólidos elegibles para trasplantes autólogos

Declaración de consenso del Reino Unido sobre el uso de plerixafor para facilitar la recolección autóloga de células madre

Acuerdos e incertidumbres en la movilización y recolección autóloga de células madre hematopoyéticas
Estudio fase I/II de dos brazos (MOZAIC): Plerixafor en pacientes pediátricos con tumores sólidos elegibles para trasplantes autólogos
Declaración de consenso del Reino Unido sobre el uso de plerixafor para facilitar la recolección autóloga de células madre
Acuerdos e incertidumbres en la movilización y recolección autóloga de células madre hematopoyéticas

Injerto tras trasplante autólogo de progenitores hematopoyéticos en pacientes movilizados con Plerixafor
Injerto tras trasplante autólogo de progenitores hematopoyéticos en pacientes movilizados con Plerixafor
Contenido mínimo de Mozobil®
PRESENTACIÓN, PRECIO Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN:
Mozobil 20 mg/ml solución inyectable - 1 vial (CN: 663769.2). PVP: 5.538,22 €. PVP IVA: 5.759,75 €. Financiado por el SNS. Medicamento sujeto a prescripción médica. Uso hospitalario.
1. Nombre del medicamento
Mozobil 20 mg/ml solución inyectable.
2. Composición cualititva y cuantitativa
Un ml de solución contiene 20 mg de plerixafor.
Cada vial contiene 24 mg de plerixafor en 1,2 ml de solución.
Excipientes con efecto conocido: Cada ml contiene aproximadamente 5 mg (0,2 mmoles) de sodio. Para consultar la lista completa de excipientes.
3. Forma farmacéutica
Solución inyectable. Solución transparente, de incolora a amarillo pálido, con un pH de 6,0-7,5 y una osmolalidad de 260-320 mOsm/kg.
TAPH, trasplante autólogo progenitores hematopoyéticos; G-CSF, factor estimulante de colonias de granulocitos; HDT, tratamiento con dosis elevada; HR: hazard ratio (cociente de riesgos instantáneos); HSC, célula madre hematopoyética; MM, mieloma múltiple; NCCN, National Comprehensive Cancer Network; LNH, linfoma no hodgkiniano; CMSP, células madre de sangre periférica; SDF-1, factor de crecimiento derivado de células estromales 1α; IMiDs, fármacos imida inmunomoduladores.
Referencias
- Ficha técnica de Mozobil
- Giralt S, Costa L, Schriber J, et al. Optimizing autologous stem cell mobilization strategies to improve patient outcomes: consensus guidelines and recommendations. Biol Blood Marrow Transplant. 2014; 20(3):295-308.
- Costa LJ, Miller AN, Alexander ET, et al. Growth factor and patient-adapted use of plerixafor is superior to CY and growth factor for autologous hematopoietic stem cells mobilization. Bone Marrow Transplant. 2011; 46(4):523-528.
- Costa LJ, Alexander ET, Hogan KR, et al. Development and validation of a decision-making algorithm to guide the use of plerixafor for autologous hematopoietic stem cell mobilization. Bone Marrow Transplant. 2011; 46(1):64-69.
- Chen J, et al. Getting blood out of a stone: Identification and management of patients with poor hematopoietic cell mobilization. Blood Reviews, 2020,100771.
- Sorigue M, et al. Relapse risk after autologous stem cell transplantation in patients with lymphoma based on CD34+ cell dose. Leukemia & Lymphoma, 2017;58(4):916-922.
- DiPersio JF, Stadtmauer EA, Nademanee A, et al.; 3102 Investigators. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood. 2009; 113(23):5720-5726.
- DiPersio JF, Micallef IN, Sti¬ PJ, et al. Phase III prospective randomized double-blindplacebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J Clin Oncol. 2009; 27(28):4767-4773.
- Lemoli RM. New strategies for stem cell mobilization. Mediterr J Hematol Infect Dis. 2012; 4(1):e2012066. doi:10.4084/MJHID.2012.066.
- Kumar SK, Callander NS, Adekola K, et al. Multiple Myeloma, Version 3.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(12):1685-1717
- Bueno JL, Alegre A, López-Villar O, et al Agreement and uncertainties in autologous haematopoietic stem cell mobilization and colletion. A Spanish consensus document. Bone Marrow Transplant. 2020;55(4):811-817.
- Shaughnessy P, Chao N, Shapiro J, et al. Pharmacoeconomics of hematopoietic stem cell mobilization: an overview of current evidence and gaps in the literature. Biol Blood Marrow Transplant. 2013; 19(9):1301-1309.
- Pusic I, Jiang SY, Landua S, et al. Impact of mobilization and remobilization strategies on achieving su cient stem cell yields for autologous transplantation. Biol Blood Marrow Transplant. 2008; 14(9):1045-1056.
- Bensinger W, DiPersio JF, McCarty JM. Improving stem cell mobilization strategies: future directions. Bone Marrow Transplant. 2009;43(3):181-195.
MAT-ES-2202717 V1 – Noviembre 2022