
Descripción del producto
Myozyme® está indicado como terapia enzimática de sustitución (TES) a largo plazo en pacientes con diagnóstico confirmado de enfermedad de Pompe (déficit de a-glucosidasa ácida). Myozyme® está indicado para pacientes adultos y pediátricos de todas las edades.

Mecanismo de acción
Myozyme® en el tratamiento de la enfermedad de Pompe infantil (IOPD)
La enfermedad de Pompe de inicio infantil (IOPD) es un trastorno multisistémico y hereditario que puede causar complicaciones respiratorias y musculares progresivas, así como muerte prematura.1,2
Sin tratamiento progresa con rapidez y casi siempre provoca la muerte antes de los 2 años de edad.1
Síntomas a los pocos meses de vida2,3
| Retraso del crecimiento | Dificultad respiratoria | Dificultad para alimentarse |
| Hipotonía | Dificultad respiratoria | Miocardiopatía |
El manejo eficaz de la enfermedad de Pompe reduce la carga de la enfermedad, estabiliza la progresión y mantiene la calidad de vida de los pacientes.4

Myozyme® modifica la evolución natural de la enfermedad de Pompe y puede utilizarse en todos los pacientes con esta enfermedad, independientemente de la edad, el sexo o los síntomas. El tratamiento temprano con Myozyme® proporciona mejores resultados clínicos, manteniendo la función motora y prolongando la supervivencia.4,5
Eficacia de Myozyme® en niños
Acción de Myozyme®
Myozyme® degrada el glucógeno lisosómico acumulado en las células del músculo esquelético y cardiaco5-8

Ensayo clínico con Myozyme®
En un ensayo clínico de 52 semanas, Myozyme® aumentó la supervivencia sin ventilación y estabilizó la función cardíaca y motora en pacientes con IOPD5,9

Estimación de Kaplan-Meier del tiempo transcurrido desde el nacimiento hasta la necesidad de ventilación mecánica o muerte.9
*Un paciente del grupo de control histórico falleción a los 44 meses de edad.
En este estudio clínico abierto, multicéntrico, multinacional, de búsqueda de dosis y de 52 semanas de duración, se examinaron la seguridad y la eficacia de MYOZYME® en 18 pacientes con enfermedad de Pompe que comenzaron el tratamiento antes de los 6 meses de edad. La eficacia se midió evaluando la supervivencia, el uso de respirador, la masa ventricular izquierda (MVI) mediante ecocardiografía, el crecimiento (peso y longitud), el desarrollo motor y el grado de discapacidad.
Los pacientes recibieron MYOZYME® en dosis de 20 mg/kg (n = 9) o 40 mg/kg (n = 9) cada 2 semanas. Los datos de supervivencia y ventilación se analizaron hasta los 18 meses de edad, en comparación con la supervivencia de un grupo control histórico de 168 pacientes con IOPD; todos los demás datos de eficacia se analizaron con respecto a los cambios desde el inicio hasta las 52 semanas de tratamiento. Los datos de seguridad fueron analizados durante todo el tratamiento.9
Acción de Myozyme®
Myozyme® degrada el glucógeno lisosómico acumulado en las células del músculo esquelético y cardiaco5-8
Ensayo clínico con Myozyme®
En un ensayo clínico de 52 semanas, Myozyme® aumentó la supervivencia sin ventilación y estabilizó la función cardíaca y motora en pacientes con IOPD5,9
Estimación de Kaplan-Meier del tiempo transcurrido desde el nacimiento hasta la necesidad de ventilación mecánica o muerte.9
*Un paciente del grupo de control histórico falleción a los 44 meses de edad.
En este estudio clínico abierto, multicéntrico, multinacional, de búsqueda de dosis y de 52 semanas de duración, se examinaron la seguridad y la eficacia de MYOZYME® en 18 pacientes con enfermedad de Pompe que comenzaron el tratamiento antes de los 6 meses de edad. La eficacia se midió evaluando la supervivencia, el uso de respirador, la masa ventricular izquierda (MVI) mediante ecocardiografía, el crecimiento (peso y longitud), el desarrollo motor y el grado de discapacidad.
Los pacientes recibieron MYOZYME® en dosis de 20 mg/kg (n = 9) o 40 mg/kg (n = 9) cada 2 semanas. Los datos de supervivencia y ventilación se analizaron hasta los 18 meses de edad, en comparación con la supervivencia de un grupo control histórico de 168 pacientes con IOPD; todos los demás datos de eficacia se analizaron con respecto a los cambios desde el inicio hasta las 52 semanas de tratamiento. Los datos de seguridad fueron analizados durante todo el tratamiento.9
Evidencia de Myozyme® en el tratamiento de IOPD
- 100% (18/18) seguía con vida y el 83% (15/18) alcanzó los 18 meses de edad, en comparación con una supervivencia del 2% (1/42) en una cohorte histórica sin tratamiento.
- 83% (15/18) no requirieron uso de soporte ventilatorio.5
- A las 104 semanas, el 63% (10/16) de los pacientes seguían sin necesitar soporte ventilatorio.5
- 100% (14/14) logró mejoras significativas en la función cardiaca, lo que derivó en reducciones de la masa ventricular izquierda (resultado mantenido durante casi 14 años).*9
*Entre otros parámetros que miden la salud cardiaca, la ecocardiografía bidireccional convencional y las mediciones de eco-Doppier convencionales se realizaron por un ecografista experimentado al inicio del estudio y a intervalos regulares a partir de entonces.
Al cabo de 1 año en un estudio clínico, los pacientes con IOPD tratados con Myozyme®:
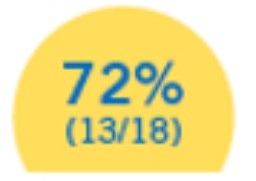
Presentaron mejoras motoras y funcionales constantes.**10
- 39% (7/18) de los pacientes pudo caminar de forma autónoma, el 17% (3/18) pudo permanecer de pie y el 17% (3/18) pudo sentarse por si mismo.10
**Las mejoras motoras se midieron utilizando la Escala Motora Infantil de Alberta.10

Mejoraron sus capacidades cognitivas, del lenguaje y sociales, aunque a un ritmo más lento que personas sanas de su misma edad.10
Después de tres años en un estudio clínico, los pacientes con IOPD tratados con Myozyme®:
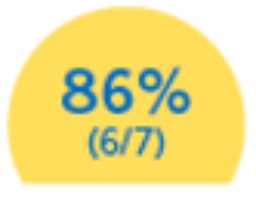
Mantuvieron su capacidad de caminar hasta el final del estudio.11
Según la última evaluación de un estudio clínico abierto, los pacientes con IOPD tratados con Myozyme® (n=21; mediana de la duración de estudio = 120 semanas):

Mejoraron su capacidad para realizar actividades de la vida diaria de forma independiente (relacionadas con la movilidad, el cuidado personal o sociales).12
En pacientes con IOPD, Myozyme®
Aumenta la supervivencia y el logro de los hitos de la función motora:5,10-13
- Prolongando la esperanza de vida
- Aumentando la supervivencia sin respirador
- Estabilizando la salud cardiaca
- Estabilizando la función motora

Tratamiento de la enfermedad de Pompe en pacientes Crim negativos
Pacientes CRIM Negativos
El tratamiento de la enfermedad de Pompe puede verse afectado negativamente por la respuesta inmune en los pacientes con enfermedad de pompe infantil clásica. Estos pacientes, denominados CRIM negativos:
- No expresan a-glucosidasa endógena
- Su sistema inmunitario jamás ha visto la proteína GAA
- En el momento en que se infunde el tratamiento, son capaces de generar anticuerpos a títulos altos.
- Dichos antiucerpos son capaces de bloquear la efectividad del tratamiento.
Generalmente, los pacientes CRIM negativos presentan una peor evolución14

Diagnóstico precoz
Hacer el diagnóstico de Pompe en Tarjeta de gota seca, DBS, facilita conocer las mutaciones del paciente, a través de una única muestra.
Predicción Genética
- El diagnóstico más habitual es encontrar 2 mutaciones nonsense mediante secuencias del gen GAA
- El estatus CRIM de cada una de las mutaciones descritas, se puede consultar en las bases de datos existentes. Ver base de datos.
Protocolos de tratamiento
Las guías españolas y britanicas de la enfermedad de Pompe de incio infantil recomiendan modular la respuesta inmunitaria de los pacientes CRIM negativos, junto con el tratamiento con la enzima recombinante15
Se ha demostrado que la respuesta inmune se asocia a la recuperación de la efectividad del farmaco, que el inicio precoz del tratamiento inmunosupresor se acompaña de una mejor respuesta terapéutica y que la combinación de inmunoglobulinas junto con el bloque de linfocitos B y T es altamente efectiva.
Es fundamental la inmunomodular al pacienteantes de poner el tratamiento con Myozyme®16
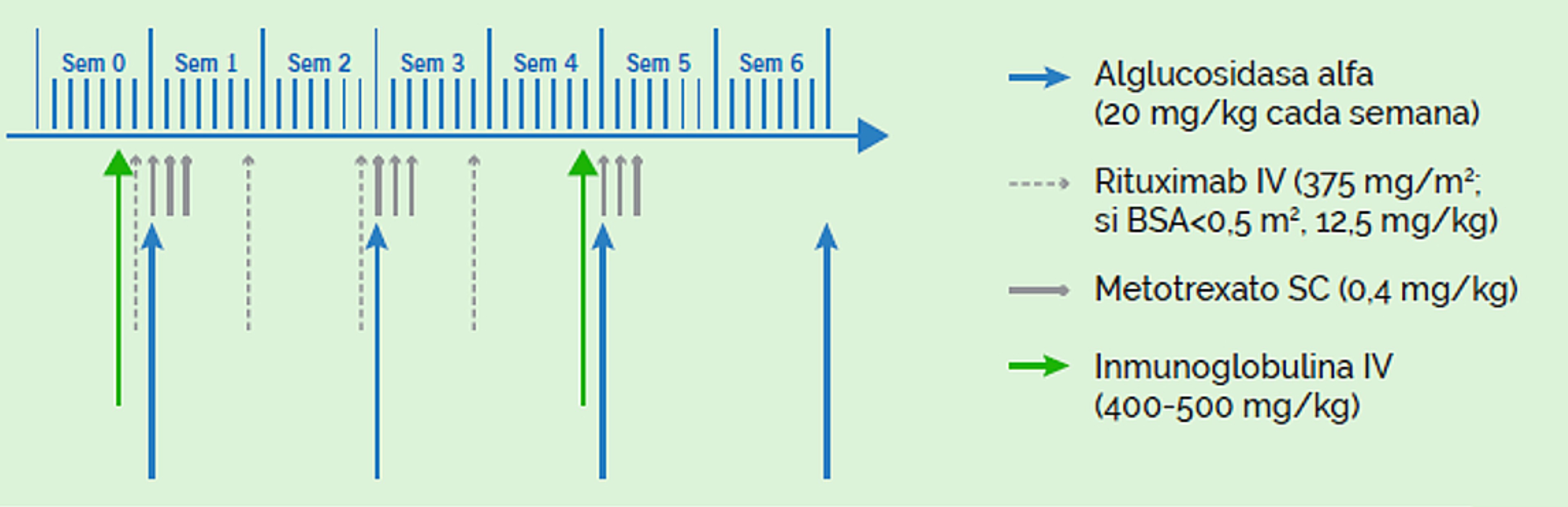
La pauta más empleada es la descrita por Banugaria SG, et al16 empleada en 7 bebés con cardiomiopatía significativa, y que consiste en la combinación de rituximab, metotrexato e inmunoglbulina IV, iniciándose dos días antes del tratamiento con Myozyme. La pauta dura 5 semanas, con administración mensual de la inmunoglobulina IV durante 5-6 meses (ver figura).
Diagnóstico precoz
Hacer el diagnóstico de Pompe en Tarjeta de gota seca, DBS, facilita conocer las mutaciones del paciente, a través de una única muestra.
Predicción Genética
- El diagnóstico más habitual es encontrar 2 mutaciones nonsense mediante secuencias del gen GAA
- El estatus CRIM de cada una de las mutaciones descritas, se puede consultar en las bases de datos existentes. Ver base de datos.
Protocolos de tratamiento
Las guías españolas y britanicas de la enfermedad de Pompe de incio infantil recomiendan modular la respuesta inmunitaria de los pacientes CRIM negativos, junto con el tratamiento con la enzima recombinante15
Se ha demostrado que la respuesta inmune se asocia a la recuperación de la efectividad del farmaco, que el inicio precoz del tratamiento inmunosupresor se acompaña de una mejor respuesta terapéutica y que la combinación de inmunoglobulinas junto con el bloque de linfocitos B y T es altamente efectiva.
Es fundamental la inmunomodular al pacienteantes de poner el tratamiento con Myozyme®16
La pauta más empleada es la descrita por Banugaria SG, et al16 empleada en 7 bebés con cardiomiopatía significativa, y que consiste en la combinación de rituximab, metotrexato e inmunoglbulina IV, iniciándose dos días antes del tratamiento con Myozyme. La pauta dura 5 semanas, con administración mensual de la inmunoglobulina IV durante 5-6 meses (ver figura).
¿Qué hacer en los casos muy graves o en bebés sintomáticos con menos de 6 meses?
"En casos muy graves, excepcionalmente se recomienda iniciar el tratamiento antes de tener los resultados del estado CRIM. En esta situación no hay datos para recomendar o desaconsejar la inmunomodulación, aunque probablemente se trate de pacientes CRIM negativos y, por tanto, podría ser aconsejable".15
"Los bebés sintomáticos con menos de 6 meses de edad tienen más probabilidades de ser CRIM negativos y deberían inciar la terapia inmunosupresiva siguiendo el protocolo para CRIM negativos".15

Conclusiones: Myozyme® en el tratamiento de la enfermedad de Pompe infantil (IOPD)
El tratamiento con TES induce la producción de anticuerpos en los pacientes con enfermedad de Pompe
Puede haber dos tipos de respuesta:

Los pacientes CRIM presentan una peor evolución y respuesta al tratamiento TES14
El tratamiento inmunomodulador permite controlar la respuesta inmunitaria con la TES15
Descarga la infografía completa de tratamiento de la enfermedad de Pompe en pacientes CRIM negativos.
Myozyme® en el tratamiento de la enfermedad de Pompe del adulto (LOPD)
Actualmente, existe un tratamiento farmacológico para la enfermedad de Pompe. Se trata de una enzima recombinante desarrollada por Sanofi Genzyme, bajo el nombre comercial de Myozyme®.
Se han realizado ensayos clínicos que abalan su utilización tanto en los casos de enfermedad de Pompe infantil como en la del adulto. Además, existen numerosas series de casos reportados en los que el tratamiento ha resultado efectivo.17
En el caso de la enfermedad de Pompe del adulto, destaca el ensayo clínico publicado en el año 2010 en la revista New England Journal of Medicine.18 Este ensayo fue un estudio aleatorizado, doble ciego y controlado con placebo que completaron 81 pacientes con enfermedad de Pompe confirmada genéticamente.
Evidencia de Myozyme® en LOPD18
En el ensayo en cuestión publicado en el New England Journal of Medicine, los criterios de inclusión aseguraban la participación en el ensayo de pacientes sintomáticos en una fase intermedia. Los criterios más relevantes eran los siguientes:
Como criterios de exclusión destacaban la necesidad de ventilación mecánica invasiva (es decir, con traqueostomía) o la necesidad de ventilación mecánica no invasiva (VNI) permanente (es decir, 24 horas al día). Si el paciente necesitaba una VNI durante la noche podía participar en el estudio.
Caminar más de 40 metros en el test de la marcha de 6 minutos (6MWT).
Tener un resultado del balance muscular obtenido mediante miómetro menor al 80% del valor.
Tener una capacidad vital forzada (CVF) en sedestación de entre un 30% y un 80% del valor.
Tener una disminución de un 10% en la CVF en posición supina respecto a la obtenida en sedestación, demostrando debilidad diafragmática.
Los pacientes fueron aleatorizados con una distribución 2:1 con placebo. 55 pacientes recibieron el tratamiento mientras que 26 recibieron placebo. La dosis de enzima recombinante fue de 20 mg/kg, administrada por vía endovenosa cada dos semanas durante 78 semanas.18
El estudio demostró diferencias significativas en el test de la marcha de 6 minutos. Los pacientes que recibían tratamiento mejoraron de media 25,13 metros, mientras que los que recibían placebo empeoraron 2,19 metros, con una diferencia total de +27,26 metros a favor del grupo tratado.18

Figura 1: Resultados del test de la marcha de 6 minutos (6MWT) en los pacientes participantes en el ensayo clínico realizado con Myozyme®. Se observan diferencias significativas entre los pacientes que recibieron tratamiento y el grupo control (Van der Ploeg AT, et al. NEJM 2010).

Figura 2: Resultados del test de la marcha de 6 minutos (6MWT) en los pacientes participantes en el ensayo clínico realizado con Myozyme®. Se observan diferencias significativas entre los pacientes que recibieron tratamiento y el grupo control (Van der Ploeg AT, et al. NEJM 2010).
La fuerza muscular se analizó mediante miometría de mano y mostró diferencias entre el grupo tratado y el que recibió placebo, pero que no fueron estadísticamente significativas. No obstante, se pudo comprobar cómo los pacientes tratados mantenían la fuerza muscular intacta respecto al inicio, mientras que en el grupo que recibía placebo había una caída de la fuerza muscular, especialmente en los músculos de las extremidades inferiores, tras 78 semanas de tratamiento.18
Los estudios de función respiratoria realizados mostraron diferencias significativas entre el grupo tratado y el de placebo. En lo referente a la CVF, se observó una mejoría del 1,2% en el grupo tratado, mientras que en el no tratado existía una pérdida del 2,2% respecto al inicio del tratamiento, lo que suponía una ganancia final del 3,4% del grupo tratado respecto a placebo (Figura 3).18

Figura 3: Evolución de la CVF de los pacientes durante las 78 semanas del ensayo clínico. Se observaron diferencias significativas entre los pacientes que recibieron tratamiento y el grupo control (Van der Ploeg AT, et al. NEJM 201018).

Figura 4: Seguimiento de pacientes con enfermedad de Pompe del adulto tratados con Myozyme®. Se observa una estabilización de los resultados tanto para la distancia recorrida en el test de la marcha de 6 minutos como para la capacidad vital forzada (van der Ploeg AT, et al. Molecular Genetics and Metabolism 201218).
Finalmente, cabe destacar la mejoría de los parámetros de presión espiratoria máxima (PEM) en los pacientes tratados (+3,24%), mientras que en el grupo control se observó una caída de la PEM (-0,56%) durante el seguimiento. Aunque los valores de presión inspiratoria máxima (PIM) se mantuvieron estables, no hubo diferencias significativas entre los grupos.18
El fármaco se toleró bien, no hubo efectos secundarios graves en ningún caso, destacando una reacción de hipersensibilidad al fármaco en dos casos (3% del grupo tratado). Todos los pacientes tratados desarrollaron anticuerpos contra la enzima, sin que hubiese relación entre el título de anticuerpos y una mala19 respuesta al tratamiento.18
Este estudio se completó con un ensayo en fase abierta.19 En este caso, se realizó seguimiento a 28 pacientes durante 130 semanas de tratamiento. Los resultados tanto del test de la marcha de 6 minutos como de la función ventilatoria mostraron una estabilidad importante, sin que se observasen variaciones destacables durante el tiempo de seguimiento (Figura 4).19
El uso de terapia de sustitución enzimática (TSE) se ha asociado de forma positiva a una mayor supervivencia, como demostró un estudio de seguimiento de 283 pacientes con enfermedad de Pompe del adulto (204 tratados y 79 no tratados) en el que se observó una reducción en la tasa de mortalidad en los pacientes que recibían la enzima recombinante. Existía una relación positiva entre la supervivencia y el tratamiento con TSE.20
Tratamiento a largo plazo con Myozyme®
Todos los datos publicados hasta el momento respecto al seguimiento de pacientes con enfermedad de Pompe por períodos prolongados de tiempo, es decir, durante varios años, proceden de estudios abiertos no aleatorizados. Estos estudios proporcionan datos de indudable interés científico, ya que nos informan de cómo evolucionan los pacientes en condiciones reales de tratamiento, pero tienen deficiencias metodológicas, debido fundamentalmente a la falta de estandarización de los métodos de seguimiento.
Se han publicado una serie de estudios abiertos de seguimiento de unos 2-3 años de pacientes con enfermedad de Pompe del adulto que mostraron una mejoría funcional, tanto motora como respiratoria, que se mantenía estable durante el tiempo de seguimiento (Figura 5).21

Figura 5: Cambios observados en la capacidad vital forzada (A) y en el test de la marcha de 6 minutos (B) a lo largo del tiempo para pacientes tratados y no tratados. Cada línea representa un estudio independiente. El tamaño de los puntos representa el tamaño aproximado de la muestra en seguimiento (Schoser B, et al. J Neurol 201721).
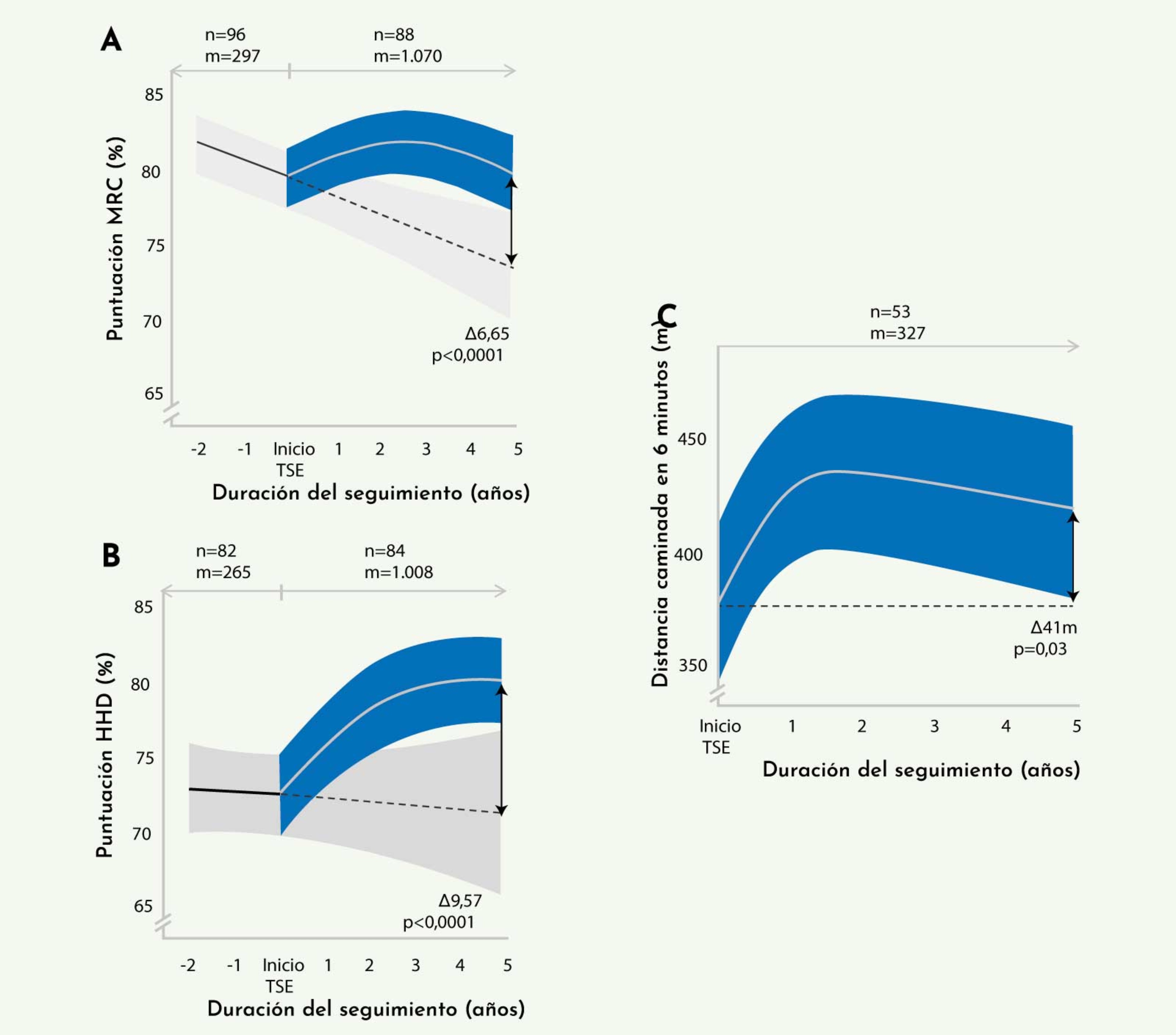
Figura 6: Cuadro de texto Figura 11. Cambios observados en la función muscular analizada mediante test manual de fuerza muscular (A), test de miometría manual (B) y test de la marcha de 6 minutos (C) (Kuperus E, et al. Neurology 201722).
En el año 2017, el grupo holandés liderado por la Dra. Van der Ploeg publicó los resultados del seguimiento de un grupo de 88 pacientes tratados por un período medio de 5 años, de los que 45 pacientes habían recibido tratamiento por más de 5 años, mientras que tan sólo 7 pacientes habían recibido tratamiento por menos de 1 año.22
El estudio mostró una mejoría inicial en la función motora de los pacientes durante los 6 primeros meses de tratamiento, valorada mediante la escala de fuerza muscular (MRC) o con la distancia recorrida en el test de la marcha de 6 minutos (Figura 6), que se mantuvo durante 2 o 3 años de seguimiento, apareciendo un posterior deterioro de la función muscular. A pesar de este deterioro final, existían diferencias significativas entre los resultados observados en los pacientes seguidos y los resultados esperados de pacientes seguidos en la época previa al tratamiento.22
Los datos mostraron que la mayoría de los pacientes seguidos tenían mejores resultados de función motora que al inicio del tratamiento, o a pesar de estar peor que al inicio, tenían mejor función motora que la esperada si no hubiesen recibido tratamiento (Figura 7).22

Figura 7: Situación funcional motora de los pacientes con enfermedad de Pompe seguidos en el estudio publicado por el grupo holandés (Kuperus E, et al. Neurology 201722).

Figura 8: Cambios observados en la función respiratoria analizada mediante capacidad vital forzada en sedestación (A), capacidad vital forzada en posición supina (B), presión inspiratoria máxima (C) y presión espiratoria máxima (D) (Kuperus E, et al. Neurology 20176).
Los resultados de la función respiratoria mostraron datos similares. Se observó una mejoría funcional, medida mediante espirometría (capacidad vital forzada (CVF) en sedestación y en posición supina, presión espiratoria máxima (PEM), y presión inspiratoria máxima (PIM)), durante los primeros 6 meses de tratamiento, que se mantenía estable por un período de 2-3 años mostrando un leve empeoramiento posterior. Al igual que ocurría con la función motora, la función respiratoria observada al final del seguimiento era mejor que la esperada por los estudios de evolución natural (Figura 8).22
Una publicación reciente analiza los datos de función respiratoria de un grupo de 396 con enfermedad de Pompe del adulto tratados con TSE y seguidos durante un período de cinco años23
Los datos se han obtenido del Pompe Registry que es un registro de pacientes con enfermedad de Pompe activo que recoge datos de forma longitudinal y está financiado y administrado por Sanofi Genzyme. Este estudio confirma los resultados positivos de la TSE sobre la función ventilatoria obtenidos en otras series de pacientes. El principal resultado es que la CVF se mantuvo estable en los pacientes del registro, tanto cuando se analizaban como un solo grupo como cuando se dividían en subgrupos, dependiendo del nivel basal de CVF (Figura 9).
Este estudio identificó además que existían diferencias en la evolución de la CVF en función del tiempo desde el diagnóstico al inicio de la TSE. Así, aquellos pacientes que habían empezado el tratamiento en un período de tiempo inferior a 1,7 años tenían una CVF mayor, de media un 3,7%, que aquellos que habían iniciado el tratamiento en un período de tiempo superior a 1,7 años, lo que sugiere un efecto beneficioso del tratamiento temprano sobre la función ventilatoria.

Figura 9: Cambios en la CVF en sedestación en un grupo de 396 pacientes con enfermedad de Pompe del adulto durante un período de seguimiento de 5 años. Los pacientes fueron divididos en 3 subgrupos en función de su CVF basal: ≥80%, >55 a <80% y ≤55%. No se observaron diferencias significativas en la CVF a lo largo del seguimiento en ninguno de los tres subgrupos (Modelo lineal general, p>0,05).

Figura 10: Diferentes patrones de progresión de los pacientes con enfermedad de Pompe del adulto tratados con TSE11
Recientemente se ha publicado un artículo que recoge datos de seguimiento de pacientes con enfermedad de Pompe tratados durante 10 años24
Se incluyó un total de 30 pacientes de los que se tenían datos prospectivos de seguimiento. Se pudo observar que existían varios tipos característicos de respuesta a la medicación como se muestra en la figura 10: pacientes en los que existía una mejoría desde el inicio que podía o no estabilizarse, pacientes que se mantenían estables con el tratamiento y pacientes en empeoraban a pesar del tratamiento. Se analizó la función motora y respiratoria mediante el test de los 6MWT y espirometría. Al igual que en los estudios realizados a los 5 años, si bien se demostró que existía un empeoramiento medio en los valores tanto del 6MWT como de la CVF en sedestación, los resultados obtenidos eran significativamente mejores a los que se habrían obtenido si se comparaba la curva de progresión de la enfermedad. El artículo mostraba que un 14% de los pacientes tenían valores de 6MWT y de CVF mejores que al inicio del tratamiento, un 38% tenía uno de los dos valores (6MWT o CVF en sedestación) mejor que al inicio del tratamiento y en un 48% de los casos, ambos valores, tanto 6MWT como CVF, eran peores que al inicio del tratamiento.
En este estudio dada la gran variabilidad interindividual, una mayoría de pacientes con LOPD se beneficia del tratamiento con TSE, al menos durante los primeros 3 a 5 años de tratamiento, seguido de un lento empeoramiento posterior por la historia natural de la enfermedad.
Cuándo empezar el tratamiento con Myozyme® en pacientes con enfermedad de Pompe del adulto (LOPD)
En el año 2014 se creó el Consorcio Europeo de la Enfermedad de Pompe (EPOC), un grupo formado por médicos e investigadores europeos interesados en esta enfermedad.25 Este consorcio publicó en el año 2017 unas guías sobre cómo realizar el seguimiento de los pacientes y cuándo empezar la terapia enzimática recombinante en los pacientes adultos.26
Estas guías recomiendan iniciar el tratamiento si se cumplen todos los supuestos siguientes:
Estas guías recomiendan iniciar el tratamiento si se cumplen todos los supuestos siguientes:
- Diagnóstico confirmado de la enfermedad de Pompe mediante la identificación de dos mutaciones en el gen GAA. En caso de que solamente se identifique una mutación, es preciso demostrar una reducción en la actividad enzimática en al menos dos tejidos distintos (leucocitos, fibroblastos de piel, biopsia muscular).
- Pacientes SINTOMÁTICOS, entendiendo como síntomas la existencia de debilidad muscular en la exploración física o la existencia de una disfunción ventilatoria en las pruebas de función
- Compromiso por parte de los pacientes para acudir a las visitas médicas pautadas y seguir los tratamientos establecidos.
- Ausencia de una enfermedad terminal que reduzca la expectativa de vida a menos de 6 meses.
Asimismo, se publicaron una serie de criterios de retirada de la medicación, siendo necesario que se cumpla uno solo de los siguientes criterios:
- Ausencia de respuesta clínica, entendiendo como tal cuando no se identifica mejoría, estabilización o cambio en la tendencia evolutiva de los pacientes tras dos años de
- Respuesta alérgica a la medicación que no es capaz de controlarse a pesar de los tratamientos
- Desarrollo de una enfermedad terminal que reduzca la esperanza de vida de los pacientes a menos de 6
- Negación de los pacientes a acudir a los controles médicos
Estas guías recomiendan iniciar el tratamiento si se cumplen todos los supuestos siguientes:
- Diagnóstico confirmado de la enfermedad de Pompe mediante la identificación de dos mutaciones en el gen GAA. En caso de que solamente se identifique una mutación, es preciso demostrar una reducción en la actividad enzimática en al menos dos tejidos distintos (leucocitos, fibroblastos de piel, biopsia muscular).
- Pacientes SINTOMÁTICOS, entendiendo como síntomas la existencia de debilidad muscular en la exploración física o la existencia de una disfunción ventilatoria en las pruebas de función
- Compromiso por parte de los pacientes para acudir a las visitas médicas pautadas y seguir los tratamientos establecidos.
- Ausencia de una enfermedad terminal que reduzca la expectativa de vida a menos de 6 meses.
Asimismo, se publicaron una serie de criterios de retirada de la medicación, siendo necesario que se cumpla uno solo de los siguientes criterios:
- Ausencia de respuesta clínica, entendiendo como tal cuando no se identifica mejoría, estabilización o cambio en la tendencia evolutiva de los pacientes tras dos años de
- Respuesta alérgica a la medicación que no es capaz de controlarse a pesar de los tratamientos
- Desarrollo de una enfermedad terminal que reduzca la esperanza de vida de los pacientes a menos de 6
- Negación de los pacientes a acudir a los controles médicos
Cuándo realizar el seguimiento de pacientes con Pompe
Debemos distinguir dos situaciones clínicas completamente distintas: pacientes sintomáticos que sigan tratamiento y pacientes presintomáticos en los que no se haya detectado ningún síntoma clínico.
Pacientes ya tratados
El objetivo del tratamiento con Myozyme® es estabilizar la función motora o enlentecer el empeoramiento de los síntomas. Como se ha descrito previamente, el tratamiento es capaz de mejorar la función motora de los pacientes durante los 6 primeros meses, pero a partir de este punto los pacientes entran en una fase de meseta en la que lo que predomina es una estabilización motora y respiratoria o un empeoramiento muy lentamente progresivo. Por lo tanto, el objetivo del tratamiento a largo plazo con Myozyme® en pacientes con enfermedad de Pompe del adulto es estabilizar la función motora. El objetivo del tratamiento con Myozyme® es estabilizar la función motora o enlentecer el empeoramiento de los síntomas. Como se ha descrito previamente, el tratamiento es capaz de mejorar la función motora de los pacientes durante los 6 primeros meses, pero a partir de este punto los pacientes entran en una fase de meseta en la que lo que predomina es una estabilización motora y respiratoria o un empeoramiento muy lentamente progresivo. Por lo tanto, el objetivo del tratamiento a largo plazo con Myozyme® en pacientes con enfermedad de Pompe del adulto es estabilizar la función motora.
Para poder establecer que el tratamiento está siendo efectivo es necesario realizar un seguimiento periódico de la función motora de los pacientes. Las guías clínicas recomiendan ver al paciente al menos una vez cada 6 meses. En estas visitas debería realizarse una entrevista clínica detallada para valorar la necesidad o no de mecanismos de ayuda para caminar (bastones, muletas, silla de ruedas…) así como una exploración física que incluya el balance muscular (MRC). Se recomienda notablemente realizar medidas funcionales que incluyan una prueba de 6MWT, asociada o no a otras pruebas funcionales como el tiempo para caminar 10 metros o el timed up&go test. Las medidas de función respiratoria son fundamentales y deben realizarse al menos una vez al año, incluyendo una espirometría en la que mediremos capacidad vital forzada en sedestación y decúbito supino. Se recomienda seguimiento en neumología para valorar las necesidades de ventilación, siendo recomendable realizar capnografía al menos en la visita inicial y posteriormente cada dos o tres años en función de la espirometría basal. El seguimiento cardiológico no debe ser tan estricto como en las formas infantiles, recomendándose una ECG anual y una ecografía del corazón en la primera visita.
Si el resultado de las pruebas muestra una estabilización clínica, podemos concluir que el tratamiento está siendo efectivo, siendo recomendable mantenerlo sin modificar la dosis. Es especialmente importante pesar al paciente para ajustar la dosis.
Si el resultado de las pruebas muestra una variación mayor al 10% se recomienda repetir las pruebas en un período de tiempo no inferior a 3 meses antes de valorar si el paciente está o no empeorando. Si confirmamos la existencia de un empeoramiento, es importante ajustar los resultados a la edad del paciente, con el objetivo de obviar aquellos empeoramientos fisiológicos. En el caso de que el ajuste muestre un empeoramiento mayor del 10%, podemos plantearnos varias opciones. La literatura científica no aclara cuál es la mejor de las medidas. Es razonable revisar el estado de los anticuerpos en sangre, si bien los estudios recientes muestran que el papel de los anticuerpos en adultos es poco o nada relevante, ya que en la mayoría de los casos son a título bajo y tienen tendencia a reducirse con el paso del tiempo.27, 28 En el improbable caso de que se detecten anticuerpos a títulos elevados, puede plantearse un tratamiento inmunosupresor con rituximab, metotrexato e inmunoglobulinas con la infusión del tratamiento, siguiendo el esquema que se realiza en formas infantiles, si bien no hay casos descritos de tratamiento de esta índole en pacientes adultos.29 En el caso de que se descarte la existencia de anticuerpos a títulos elevados se recomienda confirmar que se ha ajustado la dosis al peso del paciente. También se recomienda seguir con el tratamiento al menos durante dos años para evidenciar si el paciente continúa con esta tendencia o no.
Pacientes no tratados presintomáticos
El seguimiento de los pacientes no tratados presintomáticos varía considerablemente en función de la situación basal de los mismos. En una primera visita, se recomienda realizar una exploración física completa que incluya entrevista clínica, balance muscular (MRC), pruebas funcionales motoras (al menos 6MWT asociado al test del tiempo para caminar 10 metros o timed up&go test), espirometría completa (que incluya CVF en sedestación y decúbito), analítica general, ECG y ecocardiografía. La RM muscular de cuerpo entero con secuencias en T1 nos puede mostrar la existencia de sustitución grasa, lo que indica de forma contundente que el proceso degenerativo se ha iniciado. Los músculos que típicamente se ven afectados en pacientes presintomáticos son: musculatura paraespinal y abdominal, glúteo menor y medio y aductor mayor.30
Si todas las pruebas son normales, se recomienda visita anual del paciente. Las pruebas se repetirán en cada una de las visitas. Si existe una variación de más del 10% en los resultados, se recomienda repetir las pruebas en un período de 3 meses para confirmar la tendencia al empeoramiento. Si se confirma, se recomienda iniciar la terapia de sustitución enzimática.26
La RM muscular puede repetirse para seguir la evolución de los pacientes, si bien no está claramente establecido el momento en que debe repetirse, no teniendo sentido que se haga con una frecuencia menor a un año entre resonancias.
Pacientes ya tratados
El objetivo del tratamiento con Myozyme® es estabilizar la función motora o enlentecer el empeoramiento de los síntomas. Como se ha descrito previamente, el tratamiento es capaz de mejorar la función motora de los pacientes durante los 6 primeros meses, pero a partir de este punto los pacientes entran en una fase de meseta en la que lo que predomina es una estabilización motora y respiratoria o un empeoramiento muy lentamente progresivo. Por lo tanto, el objetivo del tratamiento a largo plazo con Myozyme® en pacientes con enfermedad de Pompe del adulto es estabilizar la función motora. El objetivo del tratamiento con Myozyme® es estabilizar la función motora o enlentecer el empeoramiento de los síntomas. Como se ha descrito previamente, el tratamiento es capaz de mejorar la función motora de los pacientes durante los 6 primeros meses, pero a partir de este punto los pacientes entran en una fase de meseta en la que lo que predomina es una estabilización motora y respiratoria o un empeoramiento muy lentamente progresivo. Por lo tanto, el objetivo del tratamiento a largo plazo con Myozyme® en pacientes con enfermedad de Pompe del adulto es estabilizar la función motora.
Para poder establecer que el tratamiento está siendo efectivo es necesario realizar un seguimiento periódico de la función motora de los pacientes. Las guías clínicas recomiendan ver al paciente al menos una vez cada 6 meses. En estas visitas debería realizarse una entrevista clínica detallada para valorar la necesidad o no de mecanismos de ayuda para caminar (bastones, muletas, silla de ruedas…) así como una exploración física que incluya el balance muscular (MRC). Se recomienda notablemente realizar medidas funcionales que incluyan una prueba de 6MWT, asociada o no a otras pruebas funcionales como el tiempo para caminar 10 metros o el timed up&go test. Las medidas de función respiratoria son fundamentales y deben realizarse al menos una vez al año, incluyendo una espirometría en la que mediremos capacidad vital forzada en sedestación y decúbito supino. Se recomienda seguimiento en neumología para valorar las necesidades de ventilación, siendo recomendable realizar capnografía al menos en la visita inicial y posteriormente cada dos o tres años en función de la espirometría basal. El seguimiento cardiológico no debe ser tan estricto como en las formas infantiles, recomendándose una ECG anual y una ecografía del corazón en la primera visita.
Si el resultado de las pruebas muestra una estabilización clínica, podemos concluir que el tratamiento está siendo efectivo, siendo recomendable mantenerlo sin modificar la dosis. Es especialmente importante pesar al paciente para ajustar la dosis.
Si el resultado de las pruebas muestra una variación mayor al 10% se recomienda repetir las pruebas en un período de tiempo no inferior a 3 meses antes de valorar si el paciente está o no empeorando. Si confirmamos la existencia de un empeoramiento, es importante ajustar los resultados a la edad del paciente, con el objetivo de obviar aquellos empeoramientos fisiológicos. En el caso de que el ajuste muestre un empeoramiento mayor del 10%, podemos plantearnos varias opciones. La literatura científica no aclara cuál es la mejor de las medidas. Es razonable revisar el estado de los anticuerpos en sangre, si bien los estudios recientes muestran que el papel de los anticuerpos en adultos es poco o nada relevante, ya que en la mayoría de los casos son a título bajo y tienen tendencia a reducirse con el paso del tiempo.27, 28 En el improbable caso de que se detecten anticuerpos a títulos elevados, puede plantearse un tratamiento inmunosupresor con rituximab, metotrexato e inmunoglobulinas con la infusión del tratamiento, siguiendo el esquema que se realiza en formas infantiles, si bien no hay casos descritos de tratamiento de esta índole en pacientes adultos.29 En el caso de que se descarte la existencia de anticuerpos a títulos elevados se recomienda confirmar que se ha ajustado la dosis al peso del paciente. También se recomienda seguir con el tratamiento al menos durante dos años para evidenciar si el paciente continúa con esta tendencia o no.
Pacientes no tratados presintomáticos
El seguimiento de los pacientes no tratados presintomáticos varía considerablemente en función de la situación basal de los mismos. En una primera visita, se recomienda realizar una exploración física completa que incluya entrevista clínica, balance muscular (MRC), pruebas funcionales motoras (al menos 6MWT asociado al test del tiempo para caminar 10 metros o timed up&go test), espirometría completa (que incluya CVF en sedestación y decúbito), analítica general, ECG y ecocardiografía. La RM muscular de cuerpo entero con secuencias en T1 nos puede mostrar la existencia de sustitución grasa, lo que indica de forma contundente que el proceso degenerativo se ha iniciado. Los músculos que típicamente se ven afectados en pacientes presintomáticos son: musculatura paraespinal y abdominal, glúteo menor y medio y aductor mayor.30
Si todas las pruebas son normales, se recomienda visita anual del paciente. Las pruebas se repetirán en cada una de las visitas. Si existe una variación de más del 10% en los resultados, se recomienda repetir las pruebas en un período de 3 meses para confirmar la tendencia al empeoramiento. Si se confirma, se recomienda iniciar la terapia de sustitución enzimática.26
La RM muscular puede repetirse para seguir la evolución de los pacientes, si bien no está claramente establecido el momento en que debe repetirse, no teniendo sentido que se haga con una frecuencia menor a un año entre resonancias.
Guías EPOC
.jpg)
Figura 11: Cómo realizar el seguimiento de un paciente con enfermedad de Pompe del adulto que ya está realizando el tratamiento de forma activa.
Descarga el dossier de valor sobre el tratamiento de la enfermedad de Pompe con Myozyme®
Contenido mínimo de Myozyme®
PRESENTACIÓN, PRECIO Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN:
Myozyme 50 mg polvo concentrado para solución para perfusión – 1 vial (CN 654213.2): PVP: 580,91 €, PVP IVA: 604,15 €. Medicamento sujeto a prescripción médica. Financiado por el SNS. Uso hospitalario.

1. Nombre del medicamento
Myozyme 50 mg polvo para concentrado para solución para perfusión.
2. Composición cualititva y cuantitativa
Un vial contiene 50 mg de alglucosidasa alfa. Después de la reconstitución, la solución contiene 5 mg de alglucosidasa alfa* por ml y después de la dilución, la concentración varía de 0,5 mg a 4 mg/ml. * La a-glucosidasa ácida humana se obtiene mediante tecnología de ADN recombinante a partir de un cultivo de células de mamíferos procedentes de ovario de hámster chino (CHO). Para consultar la lista completa de excipientes, ver sección Lista de excipientes.
3. Forma farmacéutica
Polvo para concentrado para solución para perfusión. Polvo blanco a blanquecino.
Referencias
1. Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr. 2004;144(5 suppl):S35-S43.
2. Kishnani PS, Steiner RD, Bali D, et al; ACMG Work Group on Management of Pompe Disease. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267-288.
3. Kishnani PS, Hwu WL, Mandel H, et al. Infantile-Onset Pompe Disease Natural History Study Group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148(5):671-676.
4. Barba-Romero MA, Barrot E, Bautista-Lorite J, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol. 2012;54(8):497-507.
5. Ficha técnica de MYOZYME®.
6. Van Der Ploeg AT, Carlier PG, Carlier RY, et al. Prospective exploratory muscle biopsy, imaging, and functional assessment in patients with late-onset Pompe disease treated with alglucosidase alfa: the EMBASSY study. Mol Genet Metab. 2016;119(1-2):115-123.
7. Thurberg BL, Lynch Maloney C, Vaccaro C, et al. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest. 2006;86(12):1208-1220.
8. Reuser AJ, Kroos MA, Ponne NJ, et al. Uptake and stability of human and bovine acid -glucosidase in cultured fibroblasts and skeletal muscle cells from glycogenosis type II patients. Exp Cell Res. 1984;155(1):178-189.
9. Van Capelle CI, Poelman E, Frohn-Mulder IM, et al. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int J Cardiol. 2018;269:104-110. 10. Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid -glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99-109. 11. Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alfa prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66(3):329-335. 12. Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11(3):210-219. 13. Chien YH, Lee NC, Chen CA, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. 2015;166(4):985-991.e1-2.
14. Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99(1):26-33.
15. Pascual-Pascual SI, Nascimento A, Fernández-Llamazares CM, et al. Guía clínica de la enfermedad de Pompe infantil. Rev Neurol. 2016;63(6):269-279.
16. Banugaria SG, Prater CN, Patel TT, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic metrial-negative classic infantile pompe disease: a step towards improving the efficacy of ERT-PLoS One. 2013;8(6):e67052.
17. Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol. 2013;260(4):951-9. 18. Van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362(15):1396-406.
19. Van der Ploeg AT, Barohn R, Carlson L, et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol Genet Metab. 2012;107(3):456-61. 20. Güngör D, Kruijshaar ME, Plug I, et al. Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J Rare Dis. 2013;8:49
21. Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol. 2017;264:621-30.
22. Kuperus E, Kruijshaar M, Wens S, et al. Long-term benefit of enzyme replacement therapy in Pompe disease: a 5-year prospective study. Neurology. 2017;89:2365-2373.
23. Stockton DW, Kishnani P, Van der Ploeg A, et al. Respiratory function during enzyme replacement therapy in lateonset Pompe disease: longitudinal course, prognostic factors, and the impact of time from diagnosis to treatment start. Journal of Neurology 2020.
24. Harlaar L, Hogrel JY, Perniconi B, et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology 2019; 93: 1756-1767.
25. Schoser B, Laforêt P, Kruijshaar ME, et al. European Pompe Consortium (EPOC). 208th ENMC International Workshop: Formation of a European Network to develop a European data sharing model and treatment guidelines for Pompe disease Naarden, The Netherlands, 26-28 September 2014. Neuromuscul Disord. 2015;25:674-8.
26. Van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017;24:768-e3.
27. Fernández-Simón E, Carrasco-Rozas A, Gallardo E, et al. Study of the effect of anti-rhGAA antibodies at low and intermediate titers in late onset Pompe patients treated with ERT. Mol Genet Metab. 2019;128(1- 2):129-136
28. Masat E, Laforêt P, De Antonio M, et al. Long-term exposure to Myozyme results in a decrease of antidrug antibodies in late-onset Pompe disease patients. Sci Rep. 2016;6:36182.
29. Desai AK, Li C, Rosenberg AS, et al. Immunological challenges and approaches to immunomodulation in Pompe disease: a literature review. Ann Transl Med. 2019;7(13):285.
30. Figueroa-Bonaparte S, Segovia S, Llauger J, et al. Muscle MRI Findings in Childhood/Adult Onset Pompe Disease Correlate with Muscle Function. PLoS One. 2016;11(10):e0163493.
MAT-ES-2301298 V1 Octubre 2023