
Información de prescripción
Praluent es un anticuerpo monoclonal IgG1 humano producido en células de ovario de hámster chino mediante tecnología de ADN recombinante.1 Está indicado en adultos con hipercolesterolemia primaria (familiar heterocigótica y no familiar) o dislipidemia mixta, como tratamiento complementario a la dieta: en combinación con una estatina o una estatina con otros tratamientos hipolipemiantes en pacientes que no consiguen alcanzar sus objetivos de c-LDL con la dosis máxima tolerada de una estatina o, en monoterapia o en combinación con otros tratamientos hipolipemiantes en pacientes con intolerancia a las estatinas, o en los que se contraindique el uso de una estatina.1
Asimismo, está indicado en adultos con enfermedad cardiovascular aterosclerótica establecida para reducir el riesgo cardiovascular, disminuyendo los niveles de c-LDL, como tratamiento adyuvante a la corrección de otros factores de riesgo:- en combinación con la dosis máxima tolerada de una estatina con o sin otros tratamientos hipolipemiantes o - solo o en combinación con otros tratamientos hipolipemiantes en pacientes que son intolerantes a estatinas o a los que se les ha contraindicado una estatina.1
La dosis inicial habitual de Praluent es de 75 mg administrada por vía subcutánea una vez cada 2 semanas. Los pacientes que requieran una mayor reducción del c-LDL (>60 %) pueden empezar con 150 mg una vez cada 2 semanas o 300 mg una vez cada 4 semanas (mensualmente), administrados vía subcutánea.1
La dosis de Praluent se puede individualizar en función de las características del paciente, tales como el nivel basal de c-LDL, el objetivo del tratamiento y la respuesta. Los niveles de lípidos se pueden evaluar de 4 a 8 semanas después del inicio del tratamiento o de su ajuste, y en consecuencia la dosis se puede ajustar (aumentando o disminuyendo la dosis). Si se necesita una reducción adicional del c-LDL en pacientes tratados con 75 mg una vez cada 2 semanas o 300 mg una vez cada 4 semanas (mensualmente), la dosis se puede ajustar hasta la dosis máxima de 150 mg una vez cada 2 semanas.1
Si se omite una dosis, el paciente se debe administrar la inyección lo antes posible, y a continuación, debe reanudar el tratamiento según el esquema inicial.1


de los pacientes de muy alto riesgo CV con SCA no alcanzó el objetivo de c-LDL <55 mg/dL en la primera visita de seguimiento6*†

de reducción requiere la mayoría de los pacientes con SCA para alcanzar el objetivo de c-LDL <55 mg/dL6$
Los pacientes con SCA están en riesgo inmediato de sufrir otro evento CV10,11


Añadido a estatinas de alta intensidad, Praluent® se asoció a una reducción significativa de MACE en la población total del estudio ODYSSEY OUTCOMES*2. Praluent® está indicado en adultos con enfermedad cardiovascular aterosclerótica establecida para reducir el riesgo CV, disminuyendo los niveles de c-LDL, como tratamiento adyuvante a la corrección de otros factores de riesgo3.
*Objetivo principal del estudio.**Incidencia acumulada de eventos observada en la mediana de 2,8 años.†Incidencia acumulada de eventos estimada a los 4 años.c-LDL: colesterol LDL; MACE: eventos CV adversos mayores; NNT: número necesario a tratar; RRA: reducción del riesgo absoluto.
¿Qué es lo que te preocupa? ¿Cuál es tu necesidad?
Un potente*1 iPCSK9 que cubre todas las necesidades1-5

Reducción intensiva, rápida y sostenida del c-LDL**2,3
- >62% de reducción del c-LDL
- Efecto máximo a las 4 semanas
- Sostenida hasta 4 años

La única THL más allá de las estatinas asociada a reducción de la mortalidad por todas las causas†2,3
- 15 % RRR de la mortalidad por todas las causas2

Demostrando que los pacientes alcanzan los objetivos de c-LDL actuales‡3
- 95 % de los pacientes alcanzaron el objetivo de c-LDL‡3

Perfil de seguridad establecido a largo plazo2,3

Única¥ pluma mensual para los pacientes que requieran una reducción del c-LDL >60 %3,5,6
Reducción intensiva, rápida y sostenida del c-LDL**2,3
- >62% de reducción del c-LDL
- Efecto máximo a las 4 semanas
- Sostenida hasta 4 años
La única THL más allá de las estatinas asociada a reducción de la mortalidad por todas las causas†2,3
- 15 % RRR de la mortalidad por todas las causas2
Demostrando que los pacientes alcanzan los objetivos de c-LDL actuales‡3
- 95 % de los pacientes alcanzaron el objetivo de c-LDL‡3
Perfil de seguridad establecido a largo plazo2,3
Única¥ pluma mensual para los pacientes que requieran una reducción del c-LDL >60 %3,5,6
Eficacia y Seguridad
La única THL más allá de las estatinas asociada a una de reducción de la mortalidad por cualquier causa2,3*
Reducción de la mortalidad por cualquier causa con PRALUENT® vs. placebo2,3,12

Población total
HR: 0,85 (IC 95 %: 0,73-0,98)
p = 0,0261*
Reducción del riesgo relativo mantenido a lo largo del tiempo12†

Praluent® es el único iPCSK9 asociado a una reducción de la mortalidad por cualquier causa en un estudio de eventos CV.2
El tratamiento a largo plazo con Praluent® en pacientes debidamente seleccionados puede prolongar la vida de los pacientes.12
†Solo significación estadística nominal en la evaluación jerarquizada.‡Análisis predefinido. HR: hazard ratio; IC: intervalo de confianza; iPCSK9: inhibidor de la proproteína convertasa subtilisina/kexina tipo 9; RRR: reducción del riesgo relativo.
Potente reducción del c-LDL1-3*

> 62 %
de reducción del c-LDL vs. placebo†2

4 semanas
para lograr el efecto máximo‡3

4 años
de reducción del c-LDL§2
Ha demostrado que los pacientes alcanzan el objetivo c-LDL <55mg/dl tras un SCA2*

de los pacientes con SCA alcanzó el objetivo de c-LDL <55 mg/dL con PRALUENT®2*
Añadido a estatinas de alta intensidad, Praluent® muestra un mayor beneficio en la reducción de MACE en pacientes con c-LDL basal ≥100 mg/dL*3.
*En el subgrupo de pacientes con c-LDL basal ≥100 mg/dL: mayor incidencia de MACE en el grupo de placebo y mayor RRA (valor significativo de p interacción).
Población incluida en el IPT4.c-LDL: colesterol LDL; HR: hazard ratio; IC: intervalo de confianza; IPT: Informe de Posicionamiento Terapéutico; MACE: eventos CV adversos mayores; RRA: reducción del riesgo absoluto; RRR: reducción del riesgo relativo.
Perfil de seguridad favorable a largo plazo2,3
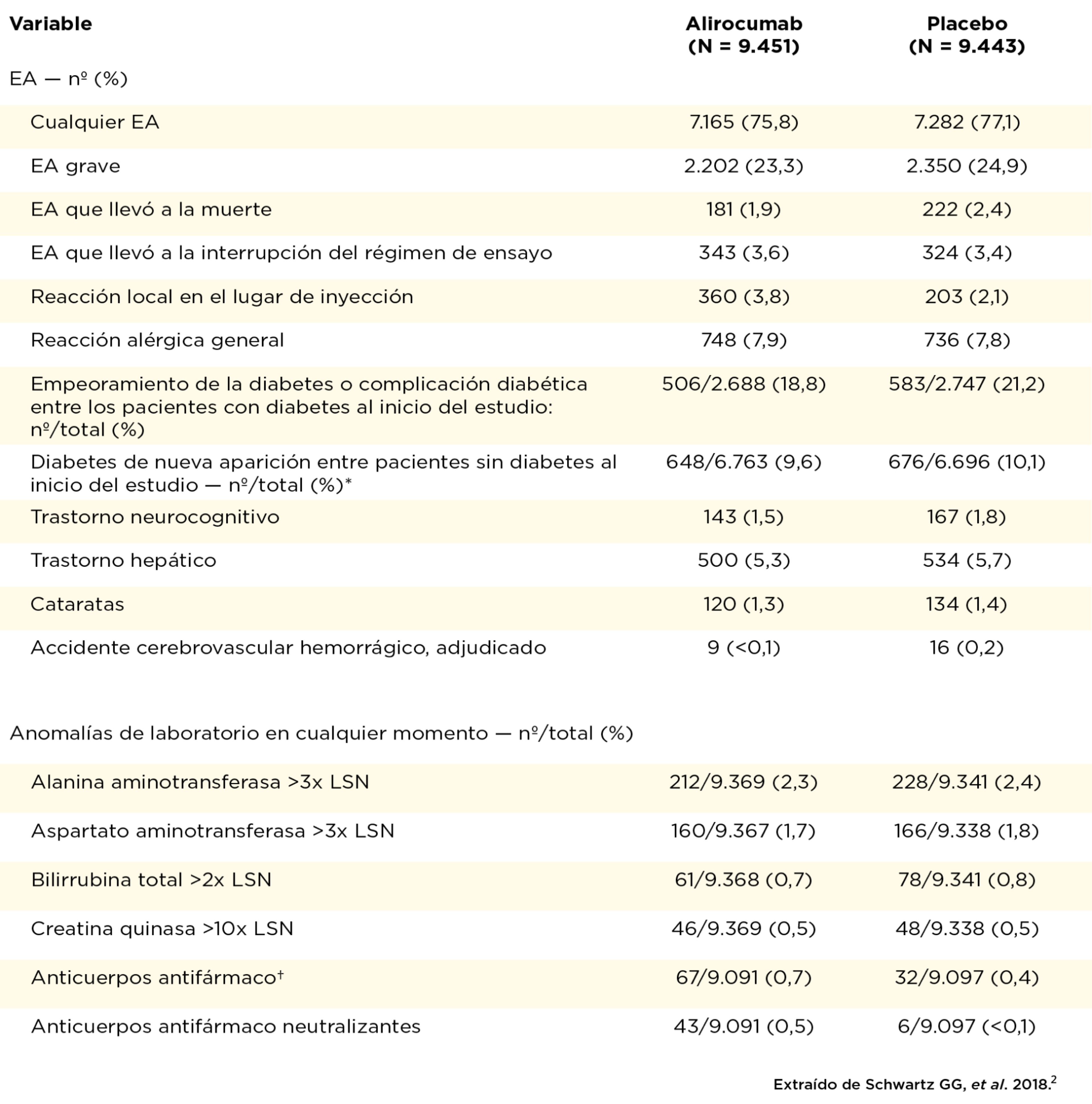
El perfil de seguridad en el estudio ODYSSEY OUTCOMES fue consistente con el perfil general de seguridad descrito en los estudios controlados de fase III. La única reacción adversa en el estudio ODYSSEY OUTCOMES que ocurrió con mayor incidencia, comparado con placebo, fue la reacción en el lugar de la inyección (p <0,001).2,3
-
Perfil de seguridad y tolerabilidad demostrado durante más de 6 años2,3.
-
El perfil de seguridad de Praluent® fue similar a placebo*2,3.
-
Praluent® reduce el riesgo de cualquier tipo de ictus e ictus isquémico, sin aumentar el riesgo de ictus hemorrágico15.
-
Praluent® no afectó negativamente a los parámetros glicémicos ni aumentó el riesgo de diabetes de nueva aparición16.
*Con la excepción de reacciones locales en el lugar de inyección (3,8 % en el grupo de Praluent® vs. 2,1 % en el de placebo + dosis máximas toleradas de estatinas, p <0,001).
EA: Efecto Adverso
Una pluma mensual* cómoda y de rápida administración para el paciente3,5

Pluma cómoda para el paciente3,5
- Aguja oculta
- Pluma sin botón de activación
- Autoadministración donde quiera que esté

Autoadministración rápida2
- Administración de la dosis ≤20 segundos2
La única THL más allá de las estatinas asociada a una de reducción de la mortalidad por cualquier causa2,3*
Reducción de la mortalidad por cualquier causa con PRALUENT® vs. placebo2,3,12
Población total
HR: 0,85 (IC 95 %: 0,73-0,98)
p = 0,0261*
Reducción del riesgo relativo mantenido a lo largo del tiempo12†
Praluent® es el único iPCSK9 asociado a una reducción de la mortalidad por cualquier causa en un estudio de eventos CV.2
El tratamiento a largo plazo con Praluent® en pacientes debidamente seleccionados puede prolongar la vida de los pacientes.12
†Solo significación estadística nominal en la evaluación jerarquizada.‡Análisis predefinido. HR: hazard ratio; IC: intervalo de confianza; iPCSK9: inhibidor de la proproteína convertasa subtilisina/kexina tipo 9; RRR: reducción del riesgo relativo.
Potente reducción del c-LDL1-3*
> 62 %
de reducción del c-LDL vs. placebo†2
4 semanas
para lograr el efecto máximo‡3
4 años
de reducción del c-LDL§2
Ha demostrado que los pacientes alcanzan el objetivo c-LDL <55mg/dl tras un SCA2*
de los pacientes con SCA alcanzó el objetivo de c-LDL <55 mg/dL con PRALUENT®2*
Añadido a estatinas de alta intensidad, Praluent® muestra un mayor beneficio en la reducción de MACE en pacientes con c-LDL basal ≥100 mg/dL*3.
*En el subgrupo de pacientes con c-LDL basal ≥100 mg/dL: mayor incidencia de MACE en el grupo de placebo y mayor RRA (valor significativo de p interacción).
Población incluida en el IPT4.c-LDL: colesterol LDL; HR: hazard ratio; IC: intervalo de confianza; IPT: Informe de Posicionamiento Terapéutico; MACE: eventos CV adversos mayores; RRA: reducción del riesgo absoluto; RRR: reducción del riesgo relativo.
Perfil de seguridad favorable a largo plazo2,3
El perfil de seguridad en el estudio ODYSSEY OUTCOMES fue consistente con el perfil general de seguridad descrito en los estudios controlados de fase III. La única reacción adversa en el estudio ODYSSEY OUTCOMES que ocurrió con mayor incidencia, comparado con placebo, fue la reacción en el lugar de la inyección (p <0,001).2,3
-
Perfil de seguridad y tolerabilidad demostrado durante más de 6 años2,3.
-
El perfil de seguridad de Praluent® fue similar a placebo*2,3.
-
Praluent® reduce el riesgo de cualquier tipo de ictus e ictus isquémico, sin aumentar el riesgo de ictus hemorrágico15.
-
Praluent® no afectó negativamente a los parámetros glicémicos ni aumentó el riesgo de diabetes de nueva aparición16.
*Con la excepción de reacciones locales en el lugar de inyección (3,8 % en el grupo de Praluent® vs. 2,1 % en el de placebo + dosis máximas toleradas de estatinas, p <0,001).
EA: Efecto Adverso
Una pluma mensual* cómoda y de rápida administración para el paciente3,5
Pluma cómoda para el paciente3,5
- Aguja oculta
- Pluma sin botón de activación
- Autoadministración donde quiera que esté
Autoadministración rápida2
- Administración de la dosis ≤20 segundos2

Pluma mensual de Praluent® 300 mg
Una pluma cómoda para el paciente1,2

Los pacientes que requieran una mayor reducción del c-LDL (>60 %) pueden empezar con 300 mg una vez cada 4 semanas (mensualmente), administrados vía subcutánea3.
La reducción del c-LDL conseguida con el dispositivo SYDNEY en la semana 4 se mantuvo durante 16 semanas5.
Los parámetros farmacocinéticos de Praluent observados con el dispositivo SYDNEY fueron similares a los obtenidos con la pluma precargada de 1 mL ya comercializada.5
Según los resultados del estudio CHOICE I†, el tratamiento mensual con Praluent 300 mg23:
- Redujo significativamente los niveles de c-LDL frente a placebo, independientemente de si el paciente estaba en tratamiento concomitante con estatinas.
- Las reducciones del c-LDL en la semana 4 se mantuvieron durante el periodo de tratamiento de 48 semanas.
Ahora Praluent en pluma precargada mensual3

Pluma precargada sin botón de activación3,5
La inyección comienza cuando el dispositivo se presiona contra la piel2. Diseñado para administrar una dosis una vez al mes en ≤20 segundos5.
Avalado por el estudio SYDNEY*, un ensayo clínico aleatorizado que demostró reducciones sustanciales del c-LDL, sin problemas técnicos asociados al dispositivo ni nuevas alertas de seguridad**5.
*Estudio multicéntrico, aleatorizado y abierto de 16 semanas para evaluar la usabilidad y las reclamaciones técnicas del producto informadas por los pacientes en un entorno sin supervisión, los acontecimientos adversos y los efectos sobre el c-LDL en pacientes que utilizan un dispositivo autoinyector de alirocumab de 2 mL (dispositivo SYDNEY). En la primera dosis, 69 pacientes con hipercolesterolemia a pesar de recibir estatinas ± otra terapia hipolipemiante recibieron de forma aleatoria alirocumab 300 mg autoadministrado con supervisión mediante una inyección de 1 × 300 mg con el dispositivo SYDNEY (n = 35) o 2 inyecciones de 150 mg con la pluma precargada ya comercializada (n = 34). Todos los pacientes que continuaron recibieron posteriormente alirocumab 300 mg cada 4 semanas autoadministrado sin supervisión utilizando el dispositivo SYDNEY en las semanas 4, 8 y 12. El criterio de valoración principal fue la proporción de reclamaciones técnicas del dispositivo SYDNEY relacionadas con el uso sin supervisión5. **En comparación con la pluma precargada de 1 mL ya comercializada5.
† Estudio aleatorizado, doble ciego, controlado con placebo, multinacional, de fase 3. El estudio comprendió un periodo de selección de 3 semanas, seguido de 48 semanas de tratamiento doble ciego y 8 semanas de seguimiento (sin tratamiento). Los pacientes fueron aleatorizados mediante un diseño de bloques permutados en una proporción de 4: 2: 1 para recibir alirocumab 300 mg cada 4 semanas, placebo o alirocumab 75 mg cada 2 semanas. La dosificación cambió en ambos grupos de alirocumab en la semana 12 si el c-LDL en la semana 8 fue ≥70 mg/dL o ≥100 mg/dL, dependiendo del riesgo CV, o si la reducción de c-LDL fue <30% desde el valor inicial en la semana 8.23
Elevada satisfacción de los pacientes2
Criterio principal de valoración: se informó de una única reclamación técnica del producto en las inyecciones sin supervisión en la semana 4 (0,5 %; 1/196 inyecciones; IC 95 %: 0,0-3,2) y se clasificó como no relacionada con el dispositivo2*

Alta adherencia al tratamiento13†
Criterio principal de valoración: PRALUENT® fue generalmente bien tolerado. Los AAET frecuentes incluyeron nasofaringitis (7,8 %), mialgia (7,1 %) y cefalea (6,2 %)14
El porcentaje de adherencia se define como el número de inyecciones recibidas dividido por el número teórico de inyecciones a recibir, multiplicado por 100.13
El nuevo dispositivo SYDNEY de 2 mL fue, en general, bien tolerado5
El perfil de seguridad fue similar al observado con Praluent 300 mg cada 4 semanas en el estudio CHOICE I 23.
No hubo problemas técnicos significativos en comparación con el dispositivo actual: se informó de una única reclamación técnica del producto en las inyecciones sin supervisión en la semana 4 (0,5%; 1/196 inyecciones; IC 95%: 0,0-3,2) y se clasificó como relacionada con el paciente y no con el dispositivo5.


# 1 paciente en el grupo del dispositivo SYDNEY.5
AAET: acontecimientos adversos emergentes con el tratamiento; IC: intervalo de confianza.
En pacientes que necesitan más protección tras un evento CV, elige Praluent.2,3



& Significación estadística nominal en la evaluación jerarquizada (HR: 0,85; IC 95%: 0,73-0,98).
§ Con la excepción de reacciones locales en el lugar de inyección (3,8% en el grupo de Praluent vs. 2,1% en el de placebo + dosis máximas toleradas de estatinas, p <0,001).
CV: cardiovascular/es; HR: hazard ratio; IC: intervalo de confianza; iPCSK9: inhibidor de PCSK9; MACE: eventos CV adversos mayores; PCSK9: proproteína convertasa subtilisina/ kexina tipo 9.
Una pluma cómoda para el paciente1,2
Los pacientes que requieran una mayor reducción del c-LDL (>60 %) pueden empezar con 300 mg una vez cada 4 semanas (mensualmente), administrados vía subcutánea3.
La reducción del c-LDL conseguida con el dispositivo SYDNEY en la semana 4 se mantuvo durante 16 semanas5.
Los parámetros farmacocinéticos de Praluent observados con el dispositivo SYDNEY fueron similares a los obtenidos con la pluma precargada de 1 mL ya comercializada.5
Según los resultados del estudio CHOICE I†, el tratamiento mensual con Praluent 300 mg23:
- Redujo significativamente los niveles de c-LDL frente a placebo, independientemente de si el paciente estaba en tratamiento concomitante con estatinas.
- Las reducciones del c-LDL en la semana 4 se mantuvieron durante el periodo de tratamiento de 48 semanas.
Ahora Praluent en pluma precargada mensual3
Pluma precargada sin botón de activación3,5
La inyección comienza cuando el dispositivo se presiona contra la piel2. Diseñado para administrar una dosis una vez al mes en ≤20 segundos5.
Avalado por el estudio SYDNEY*, un ensayo clínico aleatorizado que demostró reducciones sustanciales del c-LDL, sin problemas técnicos asociados al dispositivo ni nuevas alertas de seguridad**5.
*Estudio multicéntrico, aleatorizado y abierto de 16 semanas para evaluar la usabilidad y las reclamaciones técnicas del producto informadas por los pacientes en un entorno sin supervisión, los acontecimientos adversos y los efectos sobre el c-LDL en pacientes que utilizan un dispositivo autoinyector de alirocumab de 2 mL (dispositivo SYDNEY). En la primera dosis, 69 pacientes con hipercolesterolemia a pesar de recibir estatinas ± otra terapia hipolipemiante recibieron de forma aleatoria alirocumab 300 mg autoadministrado con supervisión mediante una inyección de 1 × 300 mg con el dispositivo SYDNEY (n = 35) o 2 inyecciones de 150 mg con la pluma precargada ya comercializada (n = 34). Todos los pacientes que continuaron recibieron posteriormente alirocumab 300 mg cada 4 semanas autoadministrado sin supervisión utilizando el dispositivo SYDNEY en las semanas 4, 8 y 12. El criterio de valoración principal fue la proporción de reclamaciones técnicas del dispositivo SYDNEY relacionadas con el uso sin supervisión5. **En comparación con la pluma precargada de 1 mL ya comercializada5.
† Estudio aleatorizado, doble ciego, controlado con placebo, multinacional, de fase 3. El estudio comprendió un periodo de selección de 3 semanas, seguido de 48 semanas de tratamiento doble ciego y 8 semanas de seguimiento (sin tratamiento). Los pacientes fueron aleatorizados mediante un diseño de bloques permutados en una proporción de 4: 2: 1 para recibir alirocumab 300 mg cada 4 semanas, placebo o alirocumab 75 mg cada 2 semanas. La dosificación cambió en ambos grupos de alirocumab en la semana 12 si el c-LDL en la semana 8 fue ≥70 mg/dL o ≥100 mg/dL, dependiendo del riesgo CV, o si la reducción de c-LDL fue <30% desde el valor inicial en la semana 8.23
Elevada satisfacción de los pacientes2
Criterio principal de valoración: se informó de una única reclamación técnica del producto en las inyecciones sin supervisión en la semana 4 (0,5 %; 1/196 inyecciones; IC 95 %: 0,0-3,2) y se clasificó como no relacionada con el dispositivo2*
Alta adherencia al tratamiento13†
Criterio principal de valoración: PRALUENT® fue generalmente bien tolerado. Los AAET frecuentes incluyeron nasofaringitis (7,8 %), mialgia (7,1 %) y cefalea (6,2 %)14
El porcentaje de adherencia se define como el número de inyecciones recibidas dividido por el número teórico de inyecciones a recibir, multiplicado por 100.13
El nuevo dispositivo SYDNEY de 2 mL fue, en general, bien tolerado5
El perfil de seguridad fue similar al observado con Praluent 300 mg cada 4 semanas en el estudio CHOICE I 23.
No hubo problemas técnicos significativos en comparación con el dispositivo actual: se informó de una única reclamación técnica del producto en las inyecciones sin supervisión en la semana 4 (0,5%; 1/196 inyecciones; IC 95%: 0,0-3,2) y se clasificó como relacionada con el paciente y no con el dispositivo5.
# 1 paciente en el grupo del dispositivo SYDNEY.5
AAET: acontecimientos adversos emergentes con el tratamiento; IC: intervalo de confianza.
En pacientes que necesitan más protección tras un evento CV, elige Praluent.2,3
& Significación estadística nominal en la evaluación jerarquizada (HR: 0,85; IC 95%: 0,73-0,98).
§ Con la excepción de reacciones locales en el lugar de inyección (3,8% en el grupo de Praluent vs. 2,1% en el de placebo + dosis máximas toleradas de estatinas, p <0,001).
CV: cardiovascular/es; HR: hazard ratio; IC: intervalo de confianza; iPCSK9: inhibidor de PCSK9; MACE: eventos CV adversos mayores; PCSK9: proproteína convertasa subtilisina/ kexina tipo 9.
Recursos Praluent 300 mg para pacientes

Contenido mínimo de Praluent®
PRESENTACIÓN, PRECIO Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN:
Praluent 75 mg pluma precargada, envase de 2 plumas precargadas (CN: 708030.5) P.V.P.: 463,06 €. Medicamento sujeto a prescripción médica.Dispensación hospitalaria.
Praluent 150 mg pluma precargada, envase de 2 plumas precargadas (CN: 708035.0) P.V.P.: 463,06 €. Medicamento sujeto a prescripción médica. Dispensación hospitalaria.
Praluent 300 mg pluma precargada, envase de 1 pluma precargada (CN: 729267.8) P.V.P.: 463,06 €. Medicamento sujeto a prescripción médica. Dispensación hospitalaria.
Ver ficha técnica Praluent 75mg
1. Nombre del medicamento
Praluent 75 mg pluma precargada, envase de 2 plumas precargadas (CN: 708030.5) P.V.P.: 463,06 €. Medicamento sujeto a prescripción médica.Dispensación hospitalaria.
Praluent 150 mg pluma precargada, envase de 2 plumas precargadas (CN: 708035.0) P.V.P.: 463,06 €. Medicamento sujeto a prescripción médica.
Dispensación hospitalaria.
Praluent 300 mg pluma precargada, envase de 1 pluma precargada (CN: 729267.8) P.V.P.: 463,06 €. Medicamento sujeto a prescripción médica.
Dispensación hospitalaria.
2. Composición cualitativa y cuantitativa
Praluent 75 mg solución inyectable en pluma precargada: Cada pluma precargada de un solo uso contiene 75 mg de alirocumab en 1 ml de solución. Praluent 150 mg solución inyectable en pluma precargada: Cada pluma precargada de un solo uso contiene 150 mg de alirocumab en 1 ml de solución. Praluent 300 mg solución inyectable en pluma precargada: Cada pluma precargada de un solo uso contiene 300 mg de alirocumab en 2 ml solución. Alirocumab es un anticuerpo monoclonal IgG1 humano producido en células de ovario de hámster chino mediante tecnología de ADN recombinante.
3. Forma farmacéutica
Solución inyectable (inyectable). Solución transparente, de incolora a amarillo pálido. pH: 5,7 - 6,3. Osmolaridad: Praluent 75 mg solución inyectable, 293 - 439 mOsm/kg. Praluent 150 mg solución inyectable, 383 - 434 mOsm/kg. Praluent 300 mg solución inyectable, 383 – 434 mOsm/kg.
AAET: acontecimientos adversos emergentes con el tratamiento. c2s: cada 2 semanas. c4s: cada 4 semanas. c-LDL: colesterol LDL. CV: cardiovascular/es. EA: evento adverso. EAS: European Atherosclerosis Society. ESC: European Society of Cardiology. HR: hazard ratio. IC: intervalo de confianza. iPCSK9: inhibidor de la proproteína convertasa subtilisina/kexina tipo 9. LSN: límite superior de la normalidad. MACE: eventos adversos cardiovasculares mayores. RRR: reducción del riesgo relativo. SCA: síndrome coronario agudo. s.c.: subcutánea. THL: terapia hipolipemiante. RRR: reducción del riesgo relativo.
HERO BANNER Y CARDS DESTACADAS INTRO
* PRALUENT®, añadido a THL máxima de base, se clasifica como terapia que permite una reducción extrema del c-LDL (76-85 %): alirocumab 75 mg (~76 %), alirocumab 150 mg (~85 %).1
** PRALUENT® redujo significativamente el riesgo de MACE (variable primaria) en la población global (N = 18.924) en el estudio ODYSSEY OUTCOMES (RRR: 15 %; HR: 0,85, IC 95 %: 0,78-0,93; p = 0,0003) y se asoció a reducción de la mortalidad por cualquier causa (RRR: 15 %; HR: 0,85, IC 95 %: 0,73-0,98, p = 0,0261), con significación estadística únicamente nominal en el análisis jerarquizado. 62,7 % de reducción comparado con placebo a los 4 meses en el estudio ODYSSEY OUTCOMES. 4 semanas para lograr el efecto máximo basado en un resumen de 10 estudios de fase 3, 5 estudios controlados con placebo y 5 estudios controlados con ezetimiba en pacientes de alto y muy alto riesgo CV. La reducción del c-LDL del 54,5 % relativo a placebo sostenida a los 4 años en pacientes tras un SCA en el estudio ODYSSEY OUTCOMES.2,3
† En un estudio de resultados CV, con significación estadística únicamente nominal en el análisis jerarquizado (HR: 0,85, IC 95 %: 0,73-0,98, p = 0,0261).2,3 ‡ Evaluación a posteriori utilizando datos del estudio ODYSSEY OUTCOMES, que incluyó a 18.924 pacientes que habían sufrido un SCA en los 12 meses previos a la inscripción y tenían c-LDL ≥70 mg/dL, niveles de colesterol no HDL ≥100 mg/dL o apoliproteína B ≥100 mg/dL a pesar del tratamiento intensivo o con dosis máxima tolerada de estatinas. Se administró a los pacientes de forma aleatoria alirocumab o placebo.2 Con PRALUENT®, el 94,6 % de los pacientes alcanzó niveles de c-LDL <1,4 mmol/L (55 mg/dL) en una o más mediciones después del basal vs. 17,3 % con placebo.4
¥ Para pacientes que requieran una reducción de c-LDL >60 %, PRALUENT® es el único iPCSK9 con una única inyección mensual en pluma precargada. 3,5,6
BANNERS 82% 60%
* Proyecto ACS EuroPath IV. El objetivo era evaluar el efecto de las guías de dislipidemia ESC/EAS 2019 en el manejo del paciente con THL en pacientes con SCA a través de una encuesta diseñada para comparar el manejo del paciente post-SCA en 2022 con el de 2018. Las guías actuales redujeron los objetivos de c-LDL recomendados para pacientes con riesgo CV muy alto de 70 mg/dL (1,8 mmol/L) a 55 mg/dL (1,4 mmol/L). Se utilizaron cuestionarios online, centrados en el perfil lipídico y la medicación para recopilar datos de 2.650 pacientes con SCA en 6 países europeos, tratados entre marzo y junio de 2022. Estos datos se compararon con los de 2.650 pacientes que participaron en la encuesta ACS EuroPath I (realizada en 2018, con metodología, criterios de reclutamiento y cuestionarios idénticos).6
† Respaldado por datos del estudio DA VINCI, un estudio observacional transversal de 18 países de 5.888 pacientes a los que se les prescribió THL para prevención primaria o secundaria en toda Europa en 2017-2018. El resultado primario fue la consecución del objetivo de c-LDL de las guías ESC/EAS 2016 basado en el riesgo mientras se recibía THL estable. También se evaluó la consecución de los objetivos de las guías del 2019, donde solo ~1/5 de los pacientes de muy alto riesgo CV alcanzó los niveles objetivo de c-LDL.2 También respaldado por datos del estudio SANTORINI, un estudio observacional y prospectivo que documentó el uso de THL de valoración primarios fueron el cambio desde el inicio en los niveles de c-LDL y el cambio desde el inicio en la utilización de THL (hasta 12 meses después de la inscripción). Entre los pacientes de muy alto riesgo CV con enfermedad aterosclerótica solo el 19,2 % alcanzó los niveles objetivo de c-LDL.8
$ Media de c-LDL al inicio del estudio: 133 mg/dL.6
EFICACIA Y SEGURIDAD Tabla 1, Pestaña 1
*Jernberg et al. realizaron un estudio de cohorte observacional y retrospectivo que analizó datos de registros nacionales suecos obligatorios. Los datos incluyeron a 97.254 pacientes ingresados en el hospital con un IM primario entre el 1 de julio de 2006 y el 30 de junio de 2011 (IM primario) y vivos 1 semana después del alta. 20.567 pacientes sufrieron un evento dentro del criterio de valoración principal compuesto de riesgo de IM no mortal, ictus no mortal o muerte por causas CV en los primeros 365 d.as posteriores al IM. La tasa acumulada del criterio de valoración principal compuesto fue del 13,3 % y el 18,3 % durante los primeros 6 y 12 meses, respectivamente, en la población con IM. Por tanto, de los eventos CV recurrentes que ocurrieron en el primer a.o después del IM, los que ocurrieron en los primeros 6 meses se han calculado como 13,3/18,3 x 100 = 72,6 %.10
† Basado en la forma de la curva de eventos compuestos, destacando la r.pida acumulación de eventos en el periodo post-IAM inmediato de 90 d.as en el estudio de Jernberg et al.1 y en los resultados de 7 ensayos intervencionistas de fase 3 diferentes en un total de 82.727 pacientes post-SCA, donde aproximadamente el 49 % de los eventos ocurrieron dentro de los primeros 90 d.as, comparado con el 51 % entre los 90 y los 360 días.11
EFICACIA Y SEGURIDAD Tabla 1: Pestaña 2
*En un estudio de resultados CV, añadida a la dosis máxima tolerada de estatinas, con significación estadística únicamente nominal en el análisis jerarquizado, p = 0,0261).2,3
† De un subanálisis de datos de mortalidad.12
¥ Análisis preespecificado.12
§ Durante el primer año, HR: 1,01 (IC 95 %: 0,77-1,32), p = 0,95. Para los pacientes elegibles para <3 años de seguimiento, HR: 0,96 (IC 95 %: 0,76-1,21), p = 0,71.12
EFICACIA Y SEGURIDAD Tabla 1: Pestaña 3
*PRALUENT®, añadido a THL máxima de base, se clasifica como terapia que permite una reducción extrema del c-LDL (76-85 %): alirocumab 75 mg (~76 %), alirocu- mab 150 mg (~85 %).1
† Reducción del 62,7 % del c-LDL en comparación con placebo a los 4 meses en el ensayo ODYSSEY OUTCOMES.2
‡ Basado en un resumen de 10 ensayos de fase 3 (5 controlados con placebo, 5 controlados con ezetimiba) en pacientes de alto y muy alto riesgo CV.3
§ Sostenida al 54,7 % a los 48 meses comparado con placebo en pacientes con un SCA en el ensayo ODYSSEY OUTCOMES.1
EFICACIA Y SEGURIDAD Tabla 1: Pestaña 4
*Diabetes de nueva aparición definida de acuerdo con la presencia de 1 o m.s de los siguientes criterios (con confirmación del diagnóstico por una revisión externa ciega por expertos en diabetes): informe de evento adverso, nueva prescripción de medicación para la diabetes, nivel de hemoglobina glicosilada de al menos 6,5 % en 2 ocasiones (y un nivel basal <6,5 %) o nivel de glucosa s.rica en ayunas de al menos 126 mg/dL (7,0 mmol/L) en 2 ocasiones (y un valor basal <126 mg/dL).2
† Los anticuerpos antifármaco se definieron por la presencia de respuestas positivas detectadas después del inicio de la administración del régimen de ensayo en al menos 2 muestras de suero postbasales consecutivas, separadas por al menos un periodo de 16 semanas.2
EFICACIA Y SEGURIDAD Tabla 1: Pestaña 5
* Para pacientes que requieren una reducción del c-LDL >60 %. La dosis inicial habitual de alirocumab es de 75 mg administrada por vía s.c. una vez cada 2 semanas. Los pacientes que requieran una mayor reducción del c-LDL (>60 %) pueden empezar con 150 mg una vez cada 2 semanas o 300 mg una vez cada 4 semanas (mensualmente), administrados por vía s.c.3
ALTA ADHERENCIA
* Estudio multicéntrico, aleatorizado, abierto, de 16 semanas en Estados Unidos. 69 pacientes con hipercolesterolemia a pesar de recibir estatinas con o sin otro tratamiento hipolipemiante recibieron aleatoriamente 300 mg de alirocumab, con supervisión durante la primera inyección, y autoadministrados a través de 1 inyección de 300 mg con el dispositivo SYDNEY (n = 35) o 2 inyecciones de 150 mg con el autoinyector inicialmente aprobado (n = 34). Los pacientes que continuaron con alirocumab recibieron posteriormente 300 mg c4s autoadministrados y sin supervisión utilizando el dispositivo SYDNEY en las semanas 4, 8 y 12. La queja técnica del producto ocurrió en la semana 4 y se clasificó como relacionada con el paciente. No se notificaron quejas técnicas del producto durante las inyecciones supervisadas.5
† ODYSSEY APPRISE fue un estudio prospectivo de fase 3b de PRALUENT® en Europa y Canadá. Este análisis post hoc examinó la adherencia del paciente al tratamiento, así como la eficacia y seguridad de PRALUENT® según el tratamiento de base con estatinas y el uso previo de ezetimiba en pacientes con hipercolesterolemia grave con riesgo CV alto. Los pacientes recibieron PRALUENT® 75 o 150 mg c2s.13
Referencias
- Escobar C, Anguita M, Arrarte V, et al. Recomendaciones para mejorar el control lipídico. Documento de consenso de la Sociedad Española de Cardiología. Rev Esp Cardiol.
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. 2020;73(2):161-167.
- Ficha técnica de PRALUENT®.
- Landmesser U, McGinniss J, Steg PG, et al. Achievement of ESC/EAS LDL-C treatment goals after an acute coronaryLsayúnndircoamTeHLwmithástaalltáindaenldas estatinas asociada alirocumab. Eur J Prev Cardiol. 2022;29(14):1842-1851.
- Frias JP, Koren MJ, Loizeau V, et al. The SYDNEY Device Study: A Multicenter, Randomized, Open-label Usability Study of a 2-mL Alirocumab Autoinjector Device. Clin Ther. 2020;42(1):94-107.
- Laufs U, Catapano AL, De Caterina R, et al. The effect of the 2019 ESC/EAS dyslipidaemia guidelines on low-density lipoprotein cholesterol goal achievement in patients with acute coronary syndromes: the ACS EuroPath IV project. Vascul Pharmacol. 2023;148:107141.
- Ray K, Molemans B, Schoonen WM, et al. EU-wide cross-sectional observational study of lipid-modifying therapy use in secondary and primary care: the DA VINCI study. Eur J Prev Cardiol. 2021;28(11):1279-1289.
- Ray K, Haq I, Bilitou A, et al. Treatment gaps in the implementation of LDL-C cholesterol control among high- and very high-risk patients in Europe between 2020 and 2021: the multinational observational SANTORINI study. Lancet Reg Health Eur. 2023;29:100624.
- Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188.
- Jernberg T, Hasvold P, Henriksson M, et al. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importan1ce of a long-term perspective. Eur Heart J. 2015;36(19):1163-1170.
- Chi G, Lee JJ, Kazmi SHA, et al. Early and late recurrent cardiovascular events among high-risk patients with an acute coronary syndrome: meta-analysis of phase 111 studies and implications on trial design. Clin Cardiol. 2022;45(3):299-307.
- Steg PG, Szarek M, Bhatt DL, et al. Effect of alirocumab on mortality after acute coronary syndromes. Circulation. 2019;140(2):103-112
- Banach M, Lopez-Sendon JL, Averna M, et al. Treatment adherence and effort of concurrent statin intensity on the efficacy and safety of alirocumab in a real-life setting: results from ODYSSEY APPRISE. Arch Med Sci. 2021;18(2):285-292.
- Gaudet D, Lopez-Sendon JL, Averna M, et al. Safety and efficacy or alirocumab in a real-life setting: the ODYSSEY APPRISE study. Eur J Prev Cardiol. 2022;28(17):1864-1872.
- Informe de Posicionamiento Terapéutico de Alirocumab (Praluent®) en hipercolesterolemia. IPT, 13/2020.V2. Disponible en: https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT_13-2020-alirocumab-Praluent.pdf?x43061. Último acceso: junio 2022.
- Schwartz GG, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097-107. Suppl. Material.
- Jukema JW, et al. Effect of alirocumab on stroke in ODYSSEY OUTCOMES. Circulation. 2019;140(25):2054-62.
- Ray KK, et al. Effects of alirocumab on cardiovascular and metabolic outcomes after acute coronary syndrome in patients with or without diabetes: a prespecified analysis of the ODYSSEY OUTCOMES randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7(8):618-28.
- Escobar C, et al. Recomendaciones para mejorar el control lipídico. Documento de consenso de la Sociedad Española de Cardiología. Rev Esp Cardiol. 2020;73:161-7.
- Arrieta F, et al. Diabetes mellitus y riesgo cardiovascular: actualización de las recomendaciones del Grupo de Trabajo de Diabetes y Enfermedad Cardiovascular de la Sociedad Española de Diabetes (SED, 2021). Clin Investig Arterioscler. 2022;34(1):36-55.
- Arrieta F, et al. Therapeutic approach with PCSK9 inhibitors for effective cardiovascular risk reduction in diabetes. Cardiol Cardiovasc Med. 2020;4(6):736-42.
- BOT PLUS. Consejo General de Colegios Farmacéuticos. Disponible en: https://botplusweb.portalfarma.com/botplus.aspx. Último acceso: agosto de 2021.
- Roth EM, Moriarty PM, Bergeron J, et al. A phase III randomized trial evaluating alirocumab 300 mg every 4 weeks as monotherapy or add-on to statin: ODYSSEY CHOICE I. Atherosclerosis. 2016;254:254-262.